Protein Structure Analysis Workflow
Jan-Philipp Quast, Patrick Stalder
2026-01-14
Source:vignettes/protein_structure_workflow.Rmd
protein_structure_workflow.RmdIntroduction
This vignette will take you through all important tools
protti provides for structural data analysis. These
include functions that allow you to fetch information about protein
structures both on an experimental as well as structural level.
Additionally, structure contact maps can be created from distances
between atoms. Lastly, B-factors* of structures can be manipulated to
contain custom values. This gives you the ability to colour regions on
structures as well as to apply continuous values associated with each
amino acid as a colour gradient using biomolecular visualisation
software such as PyMOL or ChimeraX or the R package
r3dmol.
*B-factors or temperature factors are used in structural biology to describe the atomic displacement in the structure. In low resolution structures they might correspond to the flexibility of regions, while in high resolution structures they should not be used to infer information regarding flexibility.
In structural proteomics, protein structural changes can be determined using methods such as limited proteolysis coupled to mass spectrometry (LiP-MS), cross-linking mass spectrometry (XL-MS) and hydrogen/deuterium exchange mass spectrometry (HDX-MS). In case of LiP-MS, structural changes can be identified with peptide or precursor* centric proteomics in the form of peptide or precursor fold changes. Relating this information to positions within the 3D space of a protein can be very helpful to infer potential functional roles of those structural changes. If structural changes occur close to the active site of an enzyme, kinetics might be affected, if they occur at a protein-protein binding interface, the corresponding protein complex might have dissociated or assembled.
*Peptide precursors are the different charge states of a peptide (+/- modifications) and the actual molecular unit that is detected in the mass spectrometer.
Structural data
The world wide Protein Data Bank (wwPDB) manages and archives information about the 3D structure of proteins. Its information can be accessed through several websites such as the Research Collaboratory for Structural Bioinformatics (RCSB).
When working with structure files it is important to keep a few things in mind such that mistakes can be avoided. One aspect to consider is that protein structure files come in two mainly used formats, namely the PDB file format (.pdb) and the mmCIF file format (.cif). The PDB file format is the legacy file format of the Protein Data Bank, which has been replaced with the newer mmCIF file format that stores data on biological macromolecules. All structures available in the Protein Data Bank are available as a mmCIF file, while not all are available as a PDB file. Mainly large protein complexes only contain mmCIF files.
The way both files store data and relate to each other can be quite complex. There are several resources explaining their relation and their content. The most important thing to know about these files for you as a user of protti is the fact that there are different naming conventions for elements of the structure.
PDB files solely use the naming provided by the author of the
structure in order to match the identification used in the publication
that describes the structure. Columns containing author information
start with the prefix “auth_”. This includes the naming of
chains (asym_id), atom IDs (atom_id) and
residue IDs (seq_id).
mmCIF files use both the naming provided by the author and a naming
following the standardised conventions for mmCIF files. Columns
containing standardised information start with the prefix
“label_”.
Additional information: The differences between
author provided and standardised labeling may vary a lot depending on
the structure and can be quite confusing. An example taken from the documentation
of mmCIF files describes the relationship between author provided and
standardised residue IDs as follows: The label_seq_id
(standardised residue ID) column is required to be a sequential list of
positive integers, while the auth_seq_id (author residue
ID) is not necessarily a number and the values do not have to be
positive. The author may assign values to auth_seq_id in
any desired way. For instance, the values may be used to relate this
structure to a numbering scheme in a homologous structure, including
sequence gaps or insertion codes. Alternatively, a scheme may be used
for a truncated polymer that maintains the numbering scheme of the full
length polymer. In all cases, the scheme used here must match the scheme
used in the publication that describes the structure.
For the use of the functions in protti it is
important to know that these two different conventions exist.
All functions usually take the author provided information as
inputs. This has the reason that even though the PDB file
format is a legacy format, many people still mainly use this format.
With the find_peptide_in_structure() function we provide a
convenient way for you to find both the standardised and the author
provided residue numberings for a peptide, protein region or amino acid,
returning their UniProt start and end position. In addition, we return
the full stretch of residue annotations for the author provided
information in the column auth_seq_id since they are not
necessarily consecutive as previously mentioned.
Getting started
Before starting the structural analysis of your data you need to load
protti and additional packages used for the analysis.
As described on the main page of
protti, the tidyverse package collection works
well together with the rest of the package, by design. You can load
packages with the library() function.
# Load packages
library(protti)
library(dplyr)
library(magrittr)
library(stringr)
library(tidyr)
library(ggplot2)We will demonstrate the use of protti’s structure analysis functions based on a data set of peptides that were significantly changing in a LiP-MS experiment and thus point towards a structural change in the protein.
The functions used in this analysis can also be used in a different context than a structural proteomics experiment. Instead of peptides you can investigate the positioning of individual amino acids, or whole domains within a protein.
Loading data
For this vignette we use a subset of two proteins from separate LiP-MS experiments that contain peptides significantly changing in abundance due to a structural change. We combined these two proteins into one data set to showcase that multiple proteins and structures can be analysed at once. The data was obtained from the paper “Dynamic 3D proteomes reveal protein functional alterations at high resolution in situ” (Cappelletti 2021). Both experiments were conducted on purified E. coli proteins spiked into E. coli lysates. The first protein is phosphoglycerate kinase 1 (pgk) which was treated with 25 mM 3-phosphoglyceric acid (3PG). The second protein is phosphoenolpyruvate-protein phosphotransferase (ptsI) which was treated with 25 mM fructose 1,6-bisphosphatase (FBP).
Based on structural changes observed in these proteins the authors conclude that FBP binds to the active site of ptsI and acts as a competitive inhibitor, which is confirmed by an activity assay. The structural change in pgk caused by 3PG is likely representative of substrate occupancy and correlates with metabolic flux changes.
The example data is included in protti and you can
easily use it by calling the data() function. This will
load the data into your R session.
utils::data("ptsi_pgk")If you want to read your own data into R, we suggest using the
read_protti() function. This function is a wrapper around
the fast fread() function from the data.table
package and the clean_names() function from the janitor
package. This will allow you to not only load your data into R very
fast, but also to clean up the column names into lower snake case. This
will make it easier to remember them and to use them in your data
analysis. Usually you would perform the following analysis after the
analyses described in either the vignette
for single dose treatments or the vignette
for dose response data.
# Load data
your_data <- read_protti("your_differential_abundance_data.csv")Preparing data
First we need to annotate the data with information required for
structural data analysis. To find peptides in a protein structure we
need to know their positions in the protein first. For this we can use
the protti function find_peptide(). This
function searches for the peptide within the protein sequence, which we
need to fetch from UniProt. For this we can use the
protti function fetch_uniprot(). In
addition to the protein sequence we will also need information about PDB
identifiers associated with the protein, which can also be accessed via
UniProt.
# Input UniProt IDs
uniprot_ids <- unique(ptsi_pgk$pg_protein_accessions)
# Fetch UniProt information
uniprot_information <- fetch_uniprot(
uniprot_ids = uniprot_ids,
columns = c("sequence", "xref_pdb")
)
# Add UniProt information and find peptide positions
ptsi_pgk_annotated <- ptsi_pgk %>%
left_join(uniprot_information, by = c("pg_protein_accessions" = "accession")) %>%
find_peptide(protein_sequence = sequence, peptide_sequence = pep_stripped_sequence)Fetching structural information
You can access structural information with protti in order to see if there are protein structures available for your protein, how they were acquired and if they fit your quality requirements.
In the previous step we already retrieved information about the
availability of protein structures for your protein of interest using
the fetch_uniprot() function. The column
xref_pdb contains PDB identifiers if there is a structure
available. Since the identifiers are pasted together into one string we
need to separate them into individual identifiers to provide them to the
fetch_pdb() function. This can be achieved through a
combination of the stringr function
str_split() and the tidyr function
unnest().
# Extract PDB IDs from UniProt information
ptsi_pgk_pdb_ids <- ptsi_pgk_annotated %>%
distinct(pg_protein_accessions, xref_pdb) %>%
mutate(pdb_id = str_split(xref_pdb, pattern = ";")) %>%
unnest(pdb_id) %>%
filter(pdb_id != "")
# Fetch pdb information
ptsi_pgk_pdb_information <- fetch_pdb(pdb_ids = unique(ptsi_pgk_pdb_ids$pdb_id))
#> [2/6] Extract experimental conditions ... DONE (0.02s)
#> [3/6] Extracting polymer information:
#> -> 1/7 UniProt IDs ... DONE (0.01s)
#> -> 2/7 UniProt alignment ... DONE (0.01s)
#> -> 3/7 Ligand binding sites ... DONE (0.03s)
#> -> 4/7 Modified monomers ... DONE (0.01s)
#> -> 5/7 Secondary structure ... DONE (0.01s)
#> -> 6/7 Unmodeled residues ... DONE (0.01s)
#> -> 7/7 Ligand Binding ... DONE (0s)
#> [4/6] Correct author sequence positions for some PDB IDs ... None to correct(0.01s)
#> [5/6] Extract non-polymer information ... DONE (0.01s)
#> [6/6] Combine information ... DONE (0.02s)The result of fetch_pdb() contains a multitude of
information. These include for example the experimental method that was
used to determine the structure (experimental_method and
structure_method). Depending on the method, there are
columns specific to them. For example the column
ph_crystallisation contains information about the pH of the
solution used for crystallisation if X-ray crystallography was used as a
method, while the column ph_nmr contains equivalent
information if the method was NMR. A structure resolution
(resolution_combined) is for example only included if
available, which is not the case for NMR structures.
Generally, the molecular information about the structure is divided into “polymer” and “nonpolymer”. While nonpolymer information concerns binding partners such as metal-ions, metabolites, drugs or other small molecules, the polymer information concerns the protein or polynucleotide.
You could for example only consider structures for your analysis that
were determined by X-ray crystallography and that have a resolution of
below 3 Å. In addition, you could select only the one structure that
contains the longest sequence stretch for each of our proteins (the
structure with the maximum length). This can be achieved in
the following way:
filtered_structures <- ptsi_pgk_pdb_information %>%
filter(
experimental_method == "X-ray",
resolution_combined <= 3
) %>%
group_by(reference_database_accession) %>%
filter(length == max(length)) %>%
ungroup()Generally, you can use the information retrieved by
fetch_pdb() in any way that fits your specific research
question. However, you should note that by using very strict filtering
criteria you might remove some of the proteins that actually have
structural information. This could be because their structures do not
have a high enough resolution or they are not solved using the method of
your choice. Filtering by length should also be done with care since for
some proteins there might not be any full length structure but two
structures of different domains that would make up the whole structural
information if used in combination.
Fetching atomic structural information
In addition to the general structural information that can be
retrieved with fetch_pdb() you can also access atom level
information for each structure using the
fetch_pdb_structure() function. This information is mainly
useful for applications that rely on the coordinates of each atom in the
structure. Later in this vignette we will demonstrate how this can be
used in the create_structure_contact_map() function.
Furthermore, fetch_pdb_structure() retrieves B-factors for
each atom.
Similar to fetch_pdb() PDB identifiers are provided as
input to fetch_pdb_structure(). In the example below we
provide the two PDB IDs that were left after the filtering from the
previous step was applied to the data. If not too many structures
(~<20) are fetched at once it is possible to combine all information
in a data frame by setting the return_data_frame argument
to TRUE. This is by default FALSE, which will
return a list instead with each element representing one structure.
# Input PDB IDs
pdb_ids <- unique(filtered_structures$pdb_ids) # "1ZMR", "2HWG"
# Fetch atom level structural information
ptsi_pgk_structure_information <- fetch_pdb_structure(
pdb_ids = pdb_ids,
return_data_frame = TRUE
)Fetching atomic structure prediction information from AlphaFold
If a protein does not have any available structure you can
alternatively fetch protein structure predictions from AlphaFold
using the fetch_alphafold_prediction() function. The
function works similar to fetch_pdb_structure() but it
takes UniProt identifiers instead of PDB identifiers as input.
Furthermore, there is the option to fetch all predictions for an
organism as a whole. The B-factor information in AlphaFold predictions
represents the quality score of the atom position prediction.
It is of course also possible to retrieve structure prediction
information for proteins that have a known protein structure. In the
example below we will retrieve prediction information for our two
proteins ptsi (P08839) and pgk (P0A799). The output column
score_quality annotates the score from the
prediction_score column based on the official AlphaFold
score quality annotations and cutoffs.
# Fetch atom level structural prediction information from AlphaFold
ptsi_pgk_prediction_information <- fetch_alphafold_prediction(
uniprot_ids = uniprot_ids,
return_data_frame = TRUE
)
# Example for fetching all predictions for Methanocaldococcus jannaschii
# mj_predictions <- fetch_alphafold_prediction(organism_name = "Methanocaldococcus jannaschii")AlphaFold domain predictions using the predicted aligned error (PAE)
AlphaFold predictions come with two kinds of error metrics, the per-residue confidence score (pLDDT) that is saved in the b-factor information of structure files and the predicted aligned error (PAE) that can be downloaded separately from the EBI database. According to the description on the EBI website, the PAE matrix at position (x, y) indicates AlphaFold’s expected position error at residue x, when the predicted and true structures are aligned on residue y. This is useful for assessing inter-domain accuracy. One interesting use case for the PAE is the prediction of domains in AlphaFold predictions. Tristan Croll created a python script that uses a graph-based community clustering algorithm on PAEs in order to infer pseudo-rigid protein domains. These domain predictions can be for example used in order to check the validity of distance calculations between residues of a predicted structure. If residues are on two different domains distances should be used with caution.
With protti we provide the function
fetch_alphafold_aligned_error() that allows you to retrieve
the predicted aligned error for AlphaFold predictions directly into your
R session. In addition, we adapted the python function from Tristan and
created the protti function
predict_alphafold_domain() that predicts protein domains
for AlphaFold predictions. In the example below we will retrieve PAEs
for our two proteins ptsi (P08839) and pgk (P0A799) and subsequently
predict protein domains. The error_cutoff argument of the
fetch_alphafold_aligned_error() function defines the
maximum error that should be saved. All higher values will be discarded
in order to reduce the size of the returned list. For the
predict_alphafold_domain() function we try out two
different graph_resolution values which will determine the
size of the clusters and thus how strict domains are created.
# Fetch aligned errors
aligned_error <- fetch_alphafold_aligned_error(
uniprot_ids = uniprot_ids,
error_cutoff = 4
)
# Predict protein domains with graph_resolution of 1
af_domains_res_1 <- predict_alphafold_domain(
pae_list = aligned_error,
return_data_frame = TRUE,
graph_resolution = 1
) # Default
# Predict protein domains with graph_resolution of 0.5
af_domains_res_05 <- predict_alphafold_domain(
pae_list = aligned_error,
return_data_frame = TRUE,
graph_resolution = 0.5
)Generation of structural contact maps
A very useful structural analysis function provided by
protti is create_structure_contact_map(),
which uses atom level structural information to calculate distances
between atoms or amino acid residues. The generated contact map can be
used in order to visually assess which regions are in close proximity to
each other. Furthermore, you can use this function to identify all atoms
that are in a certain proximity to a ligand or other small molecules.
Additionally, protein-protein interfaces can be easily identified in
structures that contain protein complexes.
As mentioned in the introduction it is important to find start and
end positions of e.g. peptides in a specific structure file based on the
UniProt positions. How UniProt and structure positions relate to each
other can be extracted from data fetched through
fetch_pdb(). We provide the convenient
find_peptide_in_structure() function to actually perform
the conversion of UniProt positions to structure specific positions. It
either internally uses fetch_pdb() to obtain the necessary
information or it takes the previously fetched output from
fetch_pdb() as an input to its pdb_data
argument. In the latter case it is then also possible to first filter
the data to only perform the mapping on relevant structures, which will
also speed up the process.
Note: If AlphaFold predictions are used for the contact map this step can be skipped and UniProt positions can be used directly since the AlphaFold prediction numbering is based on the UniProt positions.
In the following we are going to identify structure specific peptide
start and end positions for our annotated example data
(ptsi_pgk_annotated). By providing the previously created
filtered_structures data frame as the input to the
pdb_data argument, we will only determine the positions of
our peptides with regards to the structures contained in the filtered
data.
ptsi_pgk_peptide_structure_positions <- find_peptide_in_structure(
peptide_data = ptsi_pgk_annotated,
peptide = pep_stripped_sequence,
start = start,
end = end,
uniprot_id = pg_protein_accessions,
pdb_data = filtered_structures,
retain_columns = c(eg_precursor_id, diff, adj_pval)
)Note: The peptide argument in this function does not
necessarily require the peptide sequence but can also take any other
unique peptide identifier as input. This is possible because peptides
are not matched to the structure sequence by their sequence but are
identified based on their start and end positions in the
protein.
Before we calculate the structure contact map we are going to filter our input data to only contain significant peptides. Those are the peptides that meet our cutoff criteria for the fold change (log2(2)) and adjusted p-values (0.01). The contact map will give us an insight into regions that are in close proximity to our significant peptides. In this way we will be able to assess whether significant peptides are in close proximity to each other.
As mentioned previously, there are multiple conventions for positions
in structure files. For the create_structure_contact_map()
function it is important to provide the author defined positions and
chain names which are marked with the auth prefix. The
distance_cutoff argument is set to 10 Angstrom and will
only keep distances below that value, which will reduce the size of our
output. Since contact maps can easily reach a size of several hundred
megabytes (per map!) it is advisable to only keep the information
strictly required. Another argument that is by default set to
TRUE is return_min_residue_distance. This will
return a contact map that contains minimal residue distances instead of
atom distances, based on which the map is calculated initially.
By default contact maps are always created for the provided selection
with regards to the whole structure. If you provide an additional data
frame to the data2 argument it is possible to compare your
selection provided to the data argument to this other
subsection. This will reduce the size of contact maps if only a subset
of distances should be calculated.
Note: You can also create contact maps for AlphaFold predictions.
In that case you can provide the UniProt ID to the id
argument.
Note: If you want to create a contact map of an unpublished or
simply downloaded structure that you have the .pdb or .cif file of, you
can provide the path to that structure to the
structure_file argument. In this case the id
argument should contain a column that only has the content
“my_structure”.
Note: The input to the
create_structure_contact_map() function can be a data frame
containing specific regions (e.g. significant peptides) of a structure
that you want to create the contact map for (as explained above and
demonstrated below). It is however also possible to create contact maps
for certain chains of the structure. In that case you only need to
provide the structure ID and chain ID. Furthermore, it is possible to
generate a complete contact map of each residue in the structure. In
that case you will only need to provide the structure ID. Each of these
options will increase the size of your map and also the time it takes to
create it. Therefore, you should use these options with
caution.
# Filter data for significant peptides.
significant_peptides <- ptsi_pgk_peptide_structure_positions %>%
filter(abs(diff) > 2, adj_pval <= 0.01)
# Create a structure contact maps
contact_map <- create_structure_contact_map(
data = significant_peptides,
id = pdb_ids,
chain = auth_asym_id,
auth_seq_id = auth_seq_id,
distance_cutoff = 10,
pdb_model_number_selection = c(0, 1),
return_min_residue_distance = TRUE
)
# This is a helper function for the plot.
# It allows the display of integers on the axis.
integer_breaks <- function(n = 5, ...) {
fxn <- function(x) {
breaks <- floor(pretty(x, n, ...))
names(breaks) <- attr(breaks, "labels")
breaks
}
return(fxn)
}
# Plot structure contact maps
# 1ZMR
contact_map[["1ZMR"]] %>% # Extract data frame from list
mutate(chain_combinations = paste0("chain_", label_asym_id_var1, "_vs_chain_", label_asym_id_var2)) %>%
ggplot(aes(x = label_seq_id_var1, y = label_seq_id_var2, fill = min_distance_residue)) +
geom_tile() +
scale_y_continuous(breaks = integer_breaks()) +
scale_x_continuous(breaks = integer_breaks()) +
facet_wrap(~chain_combinations, scale = "free") +
labs(title = "Structure contact map 1ZMR") +
theme_bw()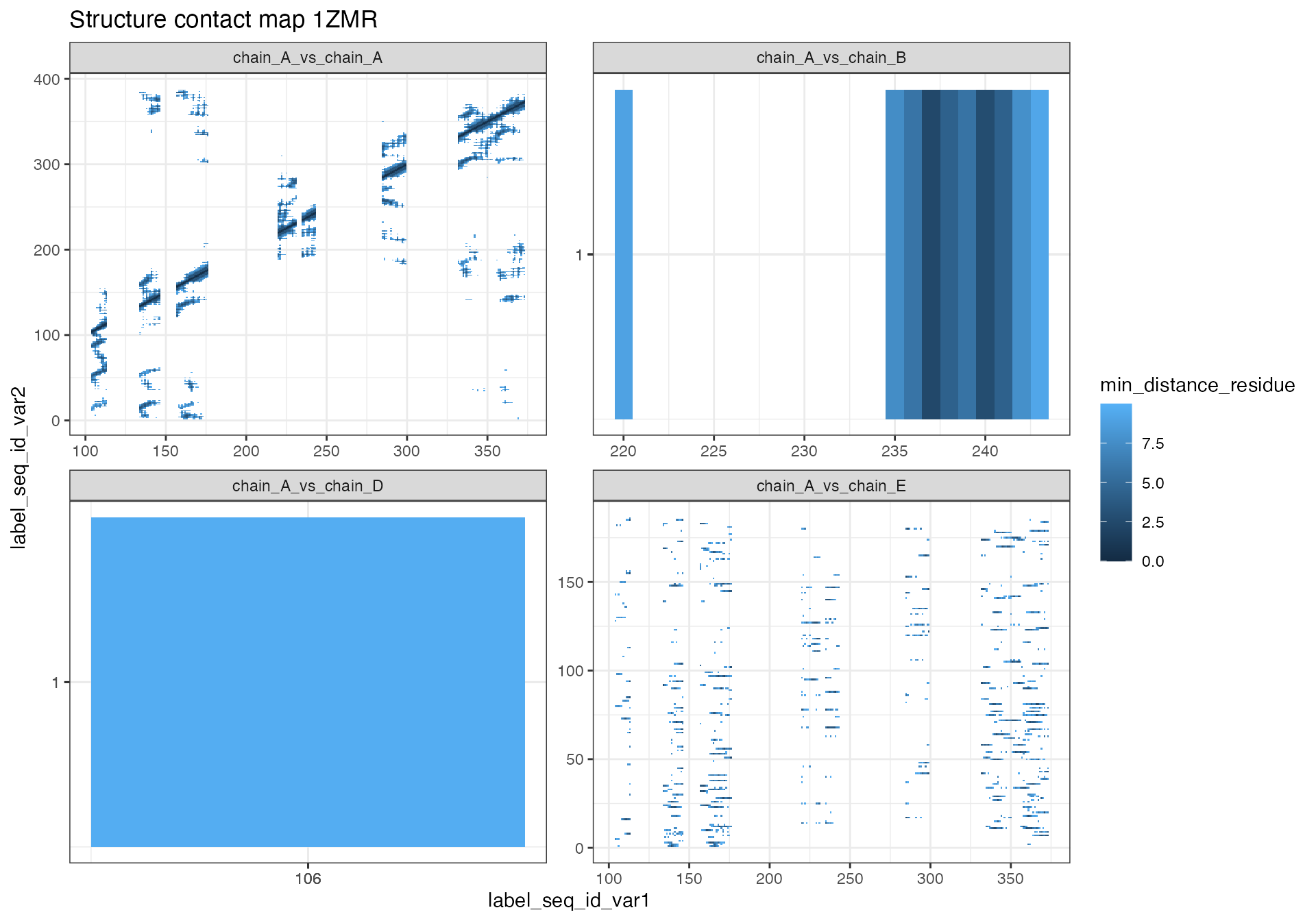
# 2HWG
contact_map[["2HWG"]] %>% # Extract data frame from list
mutate(chain_combinations = paste0("chain_", label_asym_id_var1, "_vs_chain_", label_asym_id_var2)) %>%
ggplot(aes(x = label_seq_id_var1, y = label_seq_id_var2, fill = min_distance_residue)) +
geom_tile() +
scale_y_continuous(breaks = integer_breaks()) +
scale_x_continuous(breaks = integer_breaks()) +
labs(title = "Structure contact map 2HWG") +
facet_wrap(~chain_combinations, scale = "free") +
theme_bw()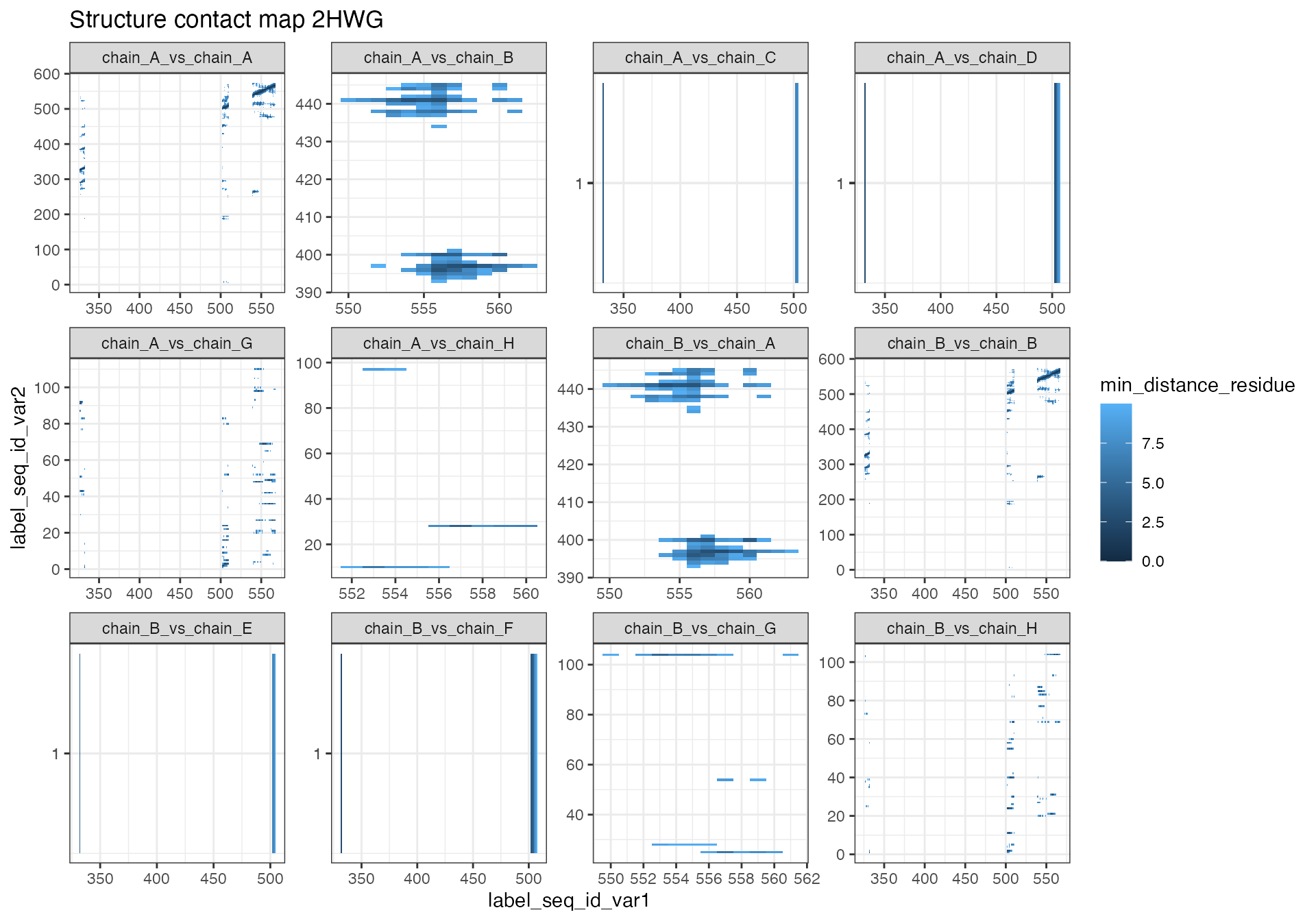 The contact map for each structure provided as input will be saved as a
separate element of a list, which is returned as the output of the
The contact map for each structure provided as input will be saved as a
separate element of a list, which is returned as the output of the
create_structure_contact_map() function. These elements can
be easily accessed using two square brackets ([[]]) right
behind the name of the list. The brackets should either contain the name
of the element or the index. You can also access each element in the
list in a loop using the map() function from the
purrr package.
Contact maps can be plotted using the ggplot2 package if
they are not too large. Contact maps are based on positions of atoms or
residues in the structure. While atom IDs are unique, residues are only
unique within a certain chain. That means residue position 1 is likely
present in each of the chains. To correctly display contact map plots
for residues they should be faceted by the unique combinations of
chains. If e.g. the x-axis displays residues from chain A and the y-axis
residues of chain B this should be displayed in one specific facet.
Note: For the plot we recommend using the data base defined chain
and residue identifiers in contrast to the author defined ones that were
used as input to the create_structure_contact_map()
function. The reason is that in the data base definitions each molecular
entity has their own chain. If a certain protein is for example
associated with a metal ion, these might be stored in the same chain in
the author defined identifiers but separated into different chains for
the data base identifiers. The problem is that in the author defined
identifiers the residue numbers might not be consecutive and there could
be a large gap in the numbering between the protein and the metal
residue.
The two contact maps that were generated based on significant peptides reveal some interesting information about the positioning of the peptides.
The “1ZMR” structure contains only one protein chain (A). In the contact map of significant peptides that are all located in chain A in relation to all other residues in chain A we can see that even though some of the peptides are quite far apart in the sequence, some are within 10 Angstrom distance in 3D space. Chain B and chain D are a calcium atom, respectively. In case of chain B the atom is actually in close proximity to one of the significant peptides. Chain E stores information about water molecules in the structure. The contact map is not very informative in this case, since the y-axis residue does not indicate any specific relationship of the water molecules with each other. Here it would be interesting to look at the distances of the water molecules to the peptide. If there are no water molecules in close proximity, this might indicate that the peptide is not surface exposed.
The “2HWG” structure contains two protein chains (A and B). Within each protein the significant peptides seem to be in close proximitiy to each other. In this case we can also observe that the peptide located around residue 555 is in close proximity to the other protein chain, which indicates that it might be located within a protein-protein interaction interface. Chain C and E contain a magnesium ion that seems to be in the proximity of two peptides. Chain D and F contain an oxalate ion which is known to inhibit ptsI. The oxalate ion is in very close proximity to to the same peptide that is close to the magnesium ion. This is likely the case because the oxalate ion and magnesium ion are located in the same site. Chain H and G contain water molecules.
Mapping of amino acids, peptides or regions on 3D protein structures and predictions
Contact maps provide quantitative information about the distances of residues to each other. We can use them to perform further high throughput analysis. In contrast to the rather complex contact map plots it is easier to look at the actual protein structures and the respective peptide positions to get a better overview of the proximity of peptides to each other and in relation to interesting sites. While this analysis is more subjective, it can make certain relationships in the data more obvious.
For the mapping of peptides, protein regions or amino acids on PDB
structures or AlphaFold predictions, we provide the
map_peptide_on_structure() function. Mapping is
accomplished based on the replacement of B-factor information in the
structure file with peptide specific values. When the structure is
coloured by B-factor in PyMOL or ChimeraX, peptides will get
highlighted. This function can on the one hand just display categorical
information such as if the peptide is significant, present or
undetected. On the other hand, continuous information can be displayed
such as a score associated with each individual amino acid.
The mapping is performed based on positions and not peptide sequence
matching. If you want to perform the mapping onto a PDB structure, you
should run the find_peptide_in_structure() beforehand to
obtain structure specific positions. If you want to map onto an
AlphaFold prediction it is not necessary to carry out this step.
However, the output of the find_peptide_in_structure()
function will contain all necessary information to perform also
AlphaFold predictions in case there is no available structure file.
For this function it is important to provide the author defined
positions and chain names which are marked with the auth
prefix. The function performs structural mapping based on this naming
convention.
Note: If you perform mapping onto an AlphaFold prediction the
content of the pdb_id provided column should be
NA.
Note: If you want to map peptides to an unpublished or downloaded
structure that you have the .pdb or .cif file of, you can provide the
path to that structure to the structure_file argument. In
this case you do not need to provide the pdb_id argument.
It is very important that your input data frame
peptide_data only contains information for the protein in
the provided structure file and not for other unrelated proteins,
otherwise mapping is not performed correctly.
You provide information on colouring through the
map_value argument. This should contain a numeric column.
In the example below we insert the number 100 into this column if a
peptide is significantly changing based on our previous cutoff criteria
and the value 0 if it is not but was detected in the experiment. In this
case you could simply insert any two numbers. However, the first number
that colours your positive hits should be higher. The function will
internally scale these values between 50 and 100. In this case 0 would
become 50 and 100 stays 100. All regions of the structure that were not
covered by any of you peptides receive the final value 0. This allows us
to distinguish between covered (50-100) and non-covered (0) regions as
well as covered non-significant (50) and significant regions (100). This
is especially helpful if peptides are coloured according to a continuous
score (see later in this vignette).
Lastly, we can specify an export location to indicate a place where
the structure file should be saved. In this example the location is
specified as a temporary file directory using the tempdir()
function. You should change that location to any more accessible place.
If you leave the argument empty, structures will be automatically saved
in your working directory.
ptsi_pgk_peptide_structure_positions %>%
mutate(map_value = ifelse(eg_precursor_id %in% significant_peptides$eg_precursor_id,
100,
0
)) %>%
map_peptides_on_structure(
uniprot_id = pg_protein_accessions,
pdb_id = pdb_ids,
chain = auth_asym_id,
auth_seq_id = auth_seq_id,
map_value = map_value,
file_format = ".pdb",
export_location = tempdir() # change to a location of your choice
)The function does not return anything in the R environment. Structures are directly saved in the desired format. The files contain the name of the structure and separated by “_” the name of all UniProt IDs present in the structure file.
To visualise coloured regions in your proteins you should open the
file in either PyMOL or ChimeraX. There you have the option to colour
structures by B-factor. Alternatively, we present an option within R
using the r3dmol package to interactively display
structures with the help of htmlwidgets.
3D structure mapping in R using r3dmol
It is possible to use the r3dmol
package to visualize protein structures directly in R. The package is
still under development and provides support for 3Dmol.js.
You can find extensive documentation and examples on their GitHub page.
Below we provide a specific example of how to use the package in order
to visualise structures created by
map_peptides_on_structure() in R colouring by B-factors.
Later we provide an example of how to use a colour gradient. In fact the
same function that is used later can also be used in this case.
If you run the code snippets on your own make sure you replace
paste0(tempdir(), "/1ZMR_P0A799.pdb") with the location and
file you want to visualise.
r3dmol facilitates direct interaction with the structure
if this vignette is exported as an html document. You can for example
zoom, rotate or move the structure.
# Install the r3dmol package if it is not installed
# install.packages("r3dmol")
# Load the r3dmol package
library(r3dmol)
# Create structure
r3dmol() %>%
m_add_model(data = paste0(tempdir(), "/1ZMR_P0A799.pdb"), format = "pdb") %>%
m_set_style(style = m_style_cartoon(
colorfunc = "
function(atom) {
if (atom.b == 50) {return '#90EE90'};
if (atom.b == 100) {return '#FF7276'};
if (atom.b == 0) {return 'white'};
return 'white';
}"
)) %>%
m_zoom_to()PyMOL
The following command colours structures by B-factor. You can specify a gradient with predefined or custom colours separating them by underscores. The gradient can also contain more than 3 different colour steps. The minimum and maximum values of the gradient should also be specified. These are based on the previously mentioned scaling. In the below gradient all non-covered regions will be gray. Covered by non-significant regions will be “palegreen” and signficiant regions will be “deepsalmon”.
spectrum b, gray70_palegreen_deepsalmon, minimum = 0, maximum = 100Custom colours can be defined with the following command and subsequently used in the command above.
Note: Custom names should not contain underscores since this confuses PyMOL when the gradient function is used.
set_color cpink, [235, 52, 189]
set_color cblue, [52, 171, 235]ChimeraX
In Chimera you have similar options like in PyMOL for colouring structures by B-factors. You can use the command below to create a colour gradient over the structure.
color bfactor palette darkgrey:darkseagreen:salmonIf you only want to colour proteins by B-factors while leaving other entities such as nucleic acids or small molecules in their default colour you can use the following command instead.
color byattribute bfactor protein palette darkgrey:darkseagreen:salmonResult
The figures below are the results of performing the colouring steps on the two structures of ptsI and pgk with significantly changing peptides highlighted in red. Areas in gray were not covered by any detected peptides.
For ptsI which was treated with fructose 1,6-bisphosphatase (FBP) the significantly changing peptides are all located in the same region of the protein. They are in close proximity to the active site that contains the magnesium atom coloured in blue and the oxalate ion coloured in purple. The contact maps showed a significant peptide in close proximity to the protein-protein interaction surface of the homodimer. This peptide is indicated by black arrows. It is not buried in the binding site but rather located at the edge of it. Its residues are, however, directly participating in the formation of the interaction surface through hydrogen bonds and likely a pi-pi stacking between a proline and histidine residue. The fact that there are no significant peptides buried in the interaction surface likely indicates that the complex does not dissociate into its subunits upon FBP treatment. It is more likely that FBP treatment induces a general structural change observable in the vicinity of the active site due to its likely binding to this site. This observation is also in line with the fact that FBP is an inhibitor of ptsI (Cappelletti 2021).
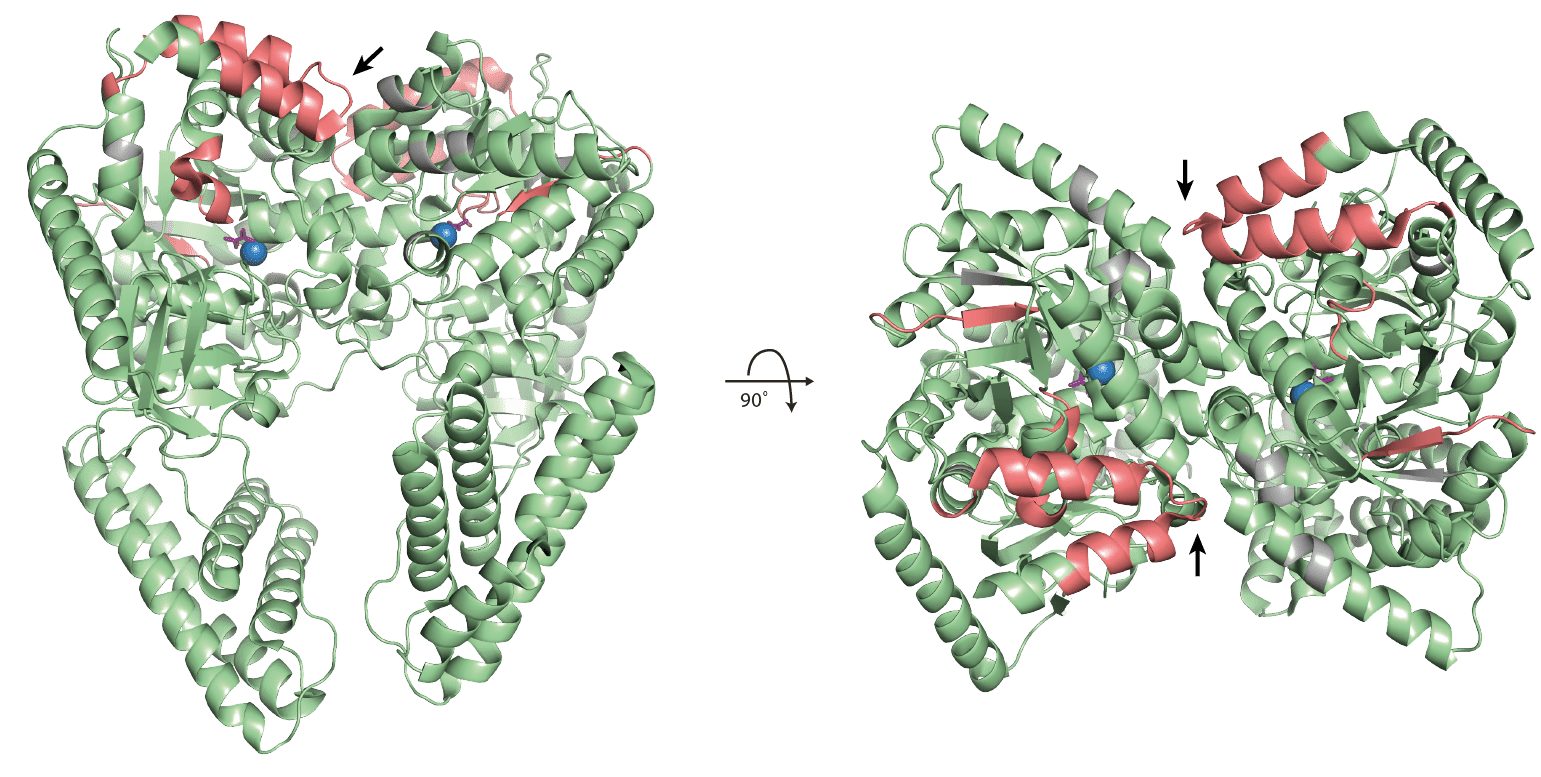
ptsI (2HWG) with mapped significantly changing peptides.
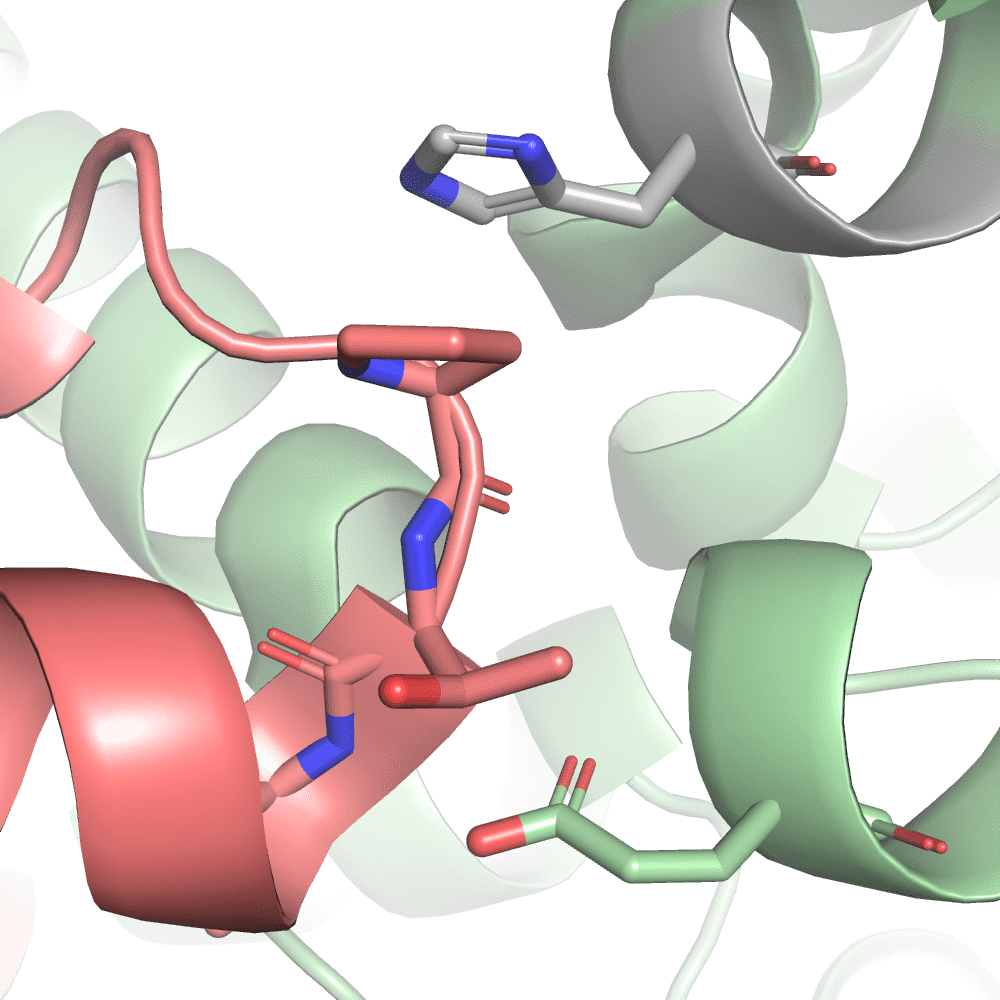
ptsI (2HWG) binding interface of significantly chaging peptide.
Mapping of significantly changing peptides onto the pgk structure reveals that most of the protein seems to be structurally affected by the treatment with 3-phosphoglyceric acid (3PG). Based on UniProt annotations and the similarity of the protein to its homologues in other organisms, the 3PG binding site is located on the left side indicated with a black arrow. None of the residues involved in the interaction with 3PG are found in a significantly changing peptide. Due to the compact size of the protein and the even distribution of significant peptides it is hard to draw any conclusions about potential structural effects of the treatment.

pgk (1ZMR) with mapped significantly changing peptides.
Mapping of amino acid scores
In order to narrow down and prioritize structurally affected protein
regions, we have developed the amino acid scoring function
calculate_aa_scores(). For each amino acid position in the
protein the function calculates the average product of the
-log10(adjusted p-value) and the absolute log2(fold change) per peptide,
covering the amino acid position. In other words, the information of all
the peptides in a specific region is integrated, which strengthens the
confidence of a structural change being true since false positive
peptides are averaged out. One downside of this approach is that it
relies on very good sequence coverage since otherwise false positive
peptides with high scores that solely cover a specific region are not
distinguishable from real hits. In our example we are lucky that both
proteins have a very high coverage.
To map the amino acid scores, the amino acid positions within the
protein structure need to be correctly assigned first using
find_peptide_in_structure(), with the residue
column as input for peptide, the start and the
end position. This step is required after the amino acid
score calculation, since calculate_aa_score() needs a
numeric input for start and end positions, and therefore,
auth_seq_id (a character column) cannot be used as an
input.
The map_peptides_on_structure() function is capable of
mapping continuous values to a structure. In this case values are again
scaled between 50 and 100. It is likely that values fall in between 50
and 100 which is displayed as a colour gradient step in between the
colour in the middle of the gradient (50) and the colour at the end of
the gradient (100).
Important: If you run this Vignette locally, make sure to save the structures generated next in a different folder than the previous ones. Otherwise the last structures will be overwritten.
Note: If multiple structures are mapped at the same time, by
default each structure scales its map values between 50 and 100 based on
its minimum and maximum score. It is also possible to use one scale for
all structures that are mapped if the scale_per_structure
argument of the map_peptides_on_structure() function is set
to FALSE. In this case only one of the structures (the one
containing the highest score) will have the 100 value. The highest score
of the other structures will be lower than 100. This means that the
scale of colours between both structures is comparable.
# Calculate the amino acid score
amino_acid_score <- calculate_aa_scores(
data = ptsi_pgk_peptide_structure_positions,
protein = pg_protein_accessions,
diff = diff,
adj_pval = adj_pval,
start_position = start,
end_position = end,
retain_columns = c(pdb_ids, auth_asym_id)
)
# Find amino acid positions in the structure
ptsi_pgk_amino_acid_structure_positions <- find_peptide_in_structure(
peptide_data = amino_acid_score,
peptide = residue,
start = residue,
end = residue,
uniprot_id = pg_protein_accessions,
pdb_data = filtered_structures,
retain_columns = c(amino_acid_score)
)
# Map the score on structure
map_peptides_on_structure(
peptide_data = ptsi_pgk_amino_acid_structure_positions,
uniprot_id = pg_protein_accessions,
pdb_id = pdb_ids,
chain = auth_asym_id,
auth_seq_id = auth_seq_id,
map_value = amino_acid_score,
file_format = ".pdb",
export_location = tempdir()
)While the previous method of displaying only significantly changing peptides that have a minimum fold change is useful for the identification of regions that change the most, it is not resistant to false positive peptides. The score on the other hand will only be high if an amino acid is consistently part of significant and differentially abundant peptides. The problem of the score is that it is hard and visually even impossible to interpret what a certain score means. This is due to the fact that the same score can be caused by two completely different scenarios. On the one hand an amino acid might be associated with peptides that have a low fold change but are highly significant. On the other hand it can be associated with peptides that have a high fold change but are of low significance. Both scenarios could yield the same amino acid score. Therefore, the score should not be misinterpreted as describing the extent of the change but should rather be seen as a probability that a certain amino acid is really structurally affected.
You can visualise the score as a colour gradient using the
r3dmol package in R. For that you need to first create a
gradient using the colorRampPalette() function, which
creates a function capable of creating a colour gradient over the
specified colours. Instead of providing only three colours to this
function you can provide additional colours to make a gradient with
multiple substeps. It is important to always use 101 colours to get the
full range for the structure. The colour gradient needs to be
concatenated using paste0() and provided to the
colorfunc argument within the shown javascript function
context.
# create a color gradient with 101 colors
color_gradient <- paste0(
'"',
paste(colorRampPalette(c("white", "#90EE90", "#FF7276"))(101),
collapse = '", "'
),
'"'
)
# create structure
r3dmol() %>%
m_add_model(data = paste0(tempdir(), "/2HWG_P08839.pdb"), format = "pdb") %>%
m_set_style(style = m_style_cartoon(
colorfunc = paste0("
function(atom) {
const color = [", color_gradient, "]
return color[Math.round(atom.b)]
}")
)) %>%
m_zoom_to()Applying the score to both structures refines the region that is most robustly affected by the treatment. For ptsI it seems that a region of amino acids directly in the active center are consistently affected. In addition we see a small region below, which was previously not seen due to its low fold changes that did not meet the cutoff. The score tells us that this region is indeed consistently affected and that there is likely a structural change occurring even though the fold change of peptides is rather low. The region in the binding interface previously indicated with a black arrow is now not highlighted anymore, indicating that this was likely a false positive hit and that other peptides in the same region do not exhibit a strong and significant response.
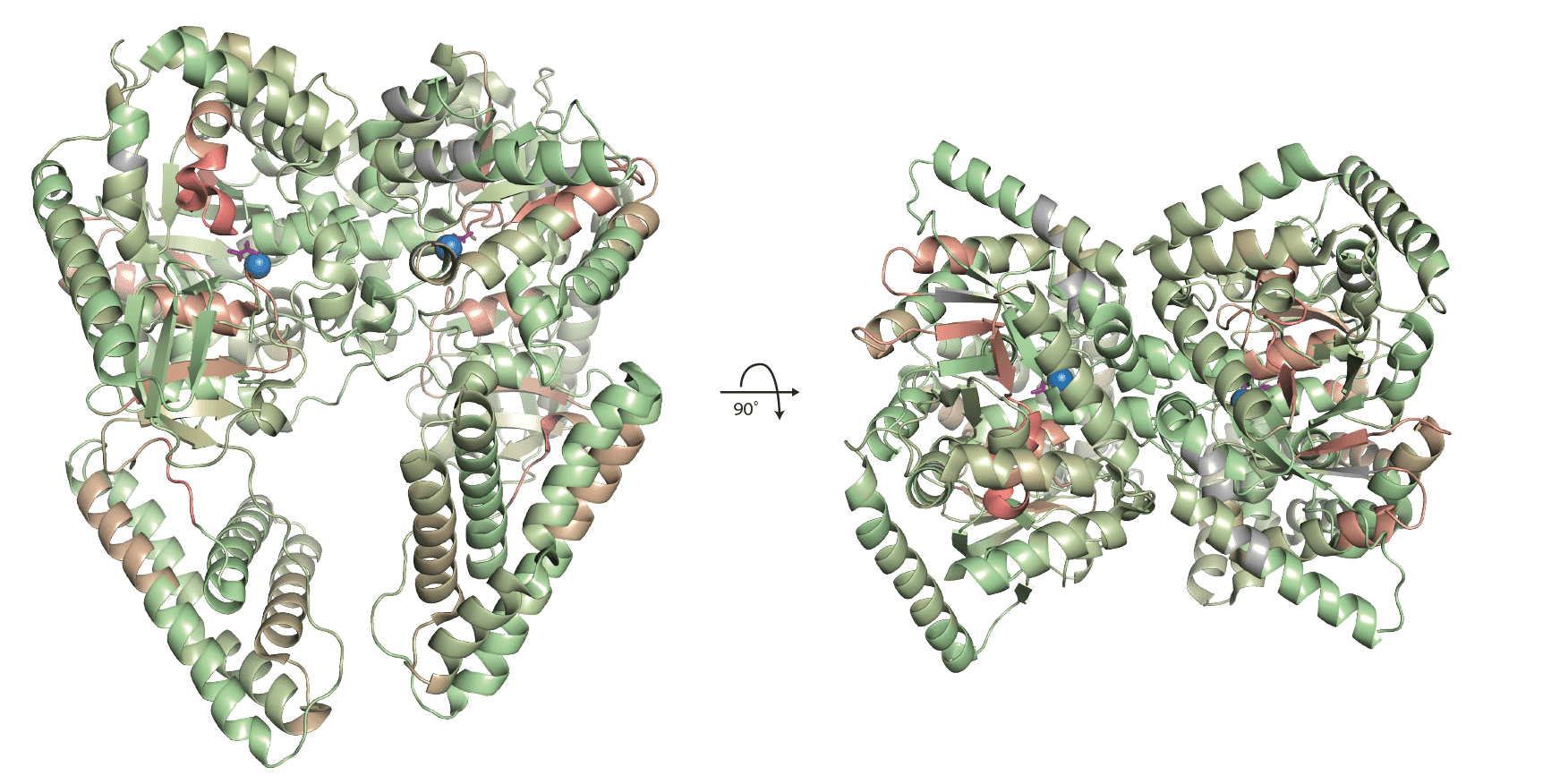
ptsI (2HWG) with mapped amino acid scores.
For ptg we see that the most robustly changing region is very close to the binding site of 3PG, which the protein was treated with. In this case, the score helped to refine the binding site of the compound since previously almost the whole protein was covered with significantly changing peptides.
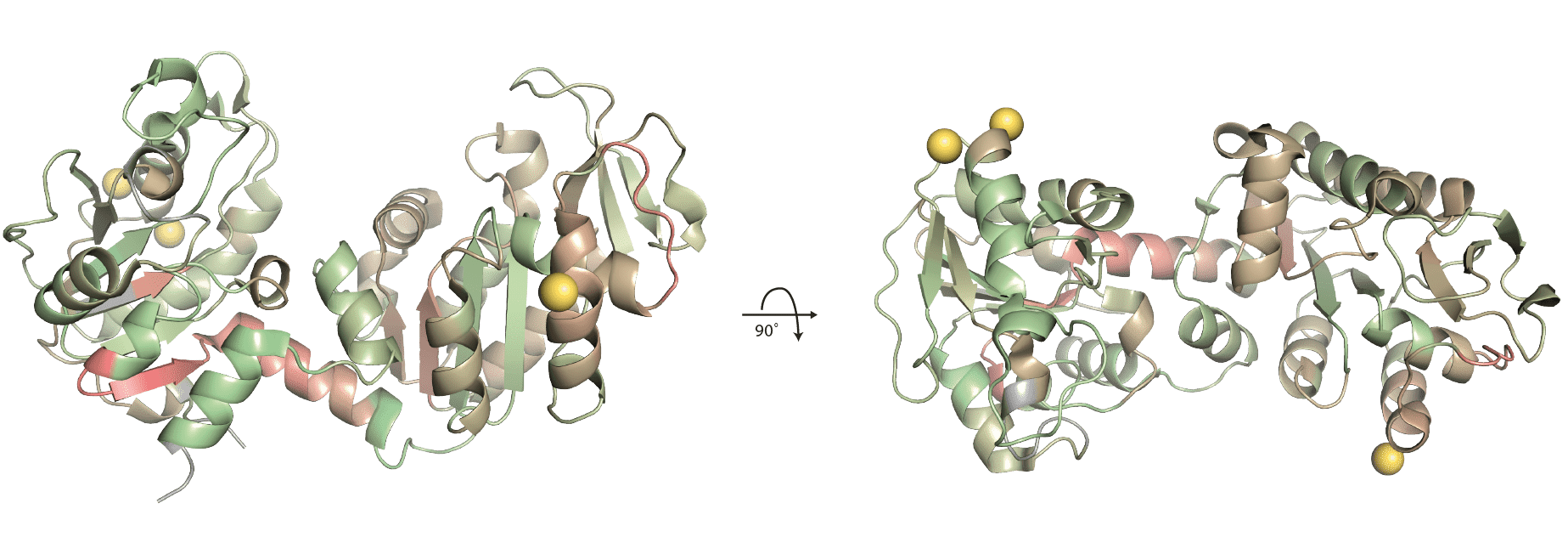
pgk (1ZMR) with mapped amino acid scores.