Qualtiy Control (QC) Workflow
Dina Schuster
2026-01-14
Source:vignettes/quality_control_workflow.Rmd
quality_control_workflow.RmdIntroduction
This vignette will give you an overview of how you can perform quality control of your bottom-up proteomics or LiP-MS data. For help with data input and to make sure that you are using the right format please check out the input preparation vignette. For help with data analysis you can check out our single dose treatment vignette and our data analysis vignette for dose-response data.
When working with proteomics and limited proteolysis-coupled mass spectrometry (LiP-MS) data, it is important to ensure that the data is of sufficient quality before inferring biological meaning from it. The goal is to assess if the data quality is homogenous among the different measurements (i.e. samples) of an experiment and if there are outliers that should be excluded.
In general, there are several ways in which this can be done:
- Inspection of .raw files acquired directly on the mass spectrometer (e.g. using vendor specific applications to visualize mass spectrometry data, such as the Thermo Scientific applications XCalibur™ and FreeStyle™)
- Different search engines and software offer quality control functionalities (e.g. Spectronaut™ offers a lot of QC options)
- R packages (e.g. PTXQC which runs on MaxQuant output or rawDiag which can be used directly on raw files)
Please note that this is not a complete list, it is just intended to give you an idea of the different possibilities. There are many more software options and R packages for quality control published to date.
protti
protti includes several functions that make it easy for the user to compare the different samples in one experiment. Some functions in this R package are specific for LiP-MS data analyses, whereas others can also be used for general proteomics analyses.
You can read more about specific functions and how to use them by
calling e.g. ?qc_cvs (for the qc_cvs()
function). Calling ? followed by the function name will
display the function documentation and give you more detailed
information about the function. This can be done for any of the
functions included in the package.
This document will give you an overview of QC functions included in
protti and will show you how they can be applied to
your data. The examples in this file are run on synthetic data, which is
created with the function create_synthetic_data().
The functions in protti are tailored towards the
analysis of DIA data, based on the output of the search engine Spectronaut™.
However, if you have any other data, such as DDA data that was searched
with a different search engine, you can still apply
protti’s functions. Just make sure that your data frame
contains tidy data.
That means data should be contained in a long format (e.g. all sample
names in one column) rather than a wide format (e.g. each sample name in
its own column). You can easily achieve this by using the
pivot_longer() function from the tidyr
package. If you are unsure about what your input data should look like,
please use the create_synthetic_data() function and compare
this to your data. You can read more about all of this here.
The input data should have a similar structure to this example:
| Sample | Replicate | Peptide Sequence | Condition | log2(Intensity) |
|---|---|---|---|---|
| sample1 | 1 | PEPTIDER | treated | 14 |
| sample1 | 1 | PEPTI | treated | 16 |
| sample1 | 1 | PEPTIDE | treated | 17 |
| sample2 | 1 | PEPTIDER | untreated | 15 |
| sample2 | 1 | PEPTI | untreated | 18 |
| sample2 | 1 | PEPTIDE | untreated | 12 |
Getting started
Before we can start analysing our data, we need to load the
protti package. This is done by using the base R
function library(). In addition, we are also loading the
packages magrittr and dplyr. Both
magrittr and dplyr are part of the tidyverse, a collection of packages
that provide useful functionalities for data processing and
visualisation. If you use many tidyverse packages in your workflow you
can easily load all at once by calling
library(tidyverse).
After having loaded the required packages we can create a synthetic dataset, that contains data similar to data obtained from a treatment experiment with e.g. a protein, metabolite or small-molecule. You can skip this step if you have your own data ready.
We are creating a random dataset with 100 different proteins, out of which 5 % are significantly changing upon treatment. The dataset includes 3 replicates for 2 different conditions (treated and untreated).
The output of the create_synthetic_data() function is
modeled after real LiP-MS data and its commonly observed data
distributions.
Please note that generally, quality control should be conducted on raw unfiltered data (the direct output of your search engine of choice).
# by setting the seed we are making sure that the random object generation can be reproduced
set.seed(123)
data <- create_synthetic_data(
n_proteins = 100,
frac_change = 0.05,
n_replicates = 3,
n_conditions = 2,
method = "effect_random",
additional_metadata = TRUE
)Quality control
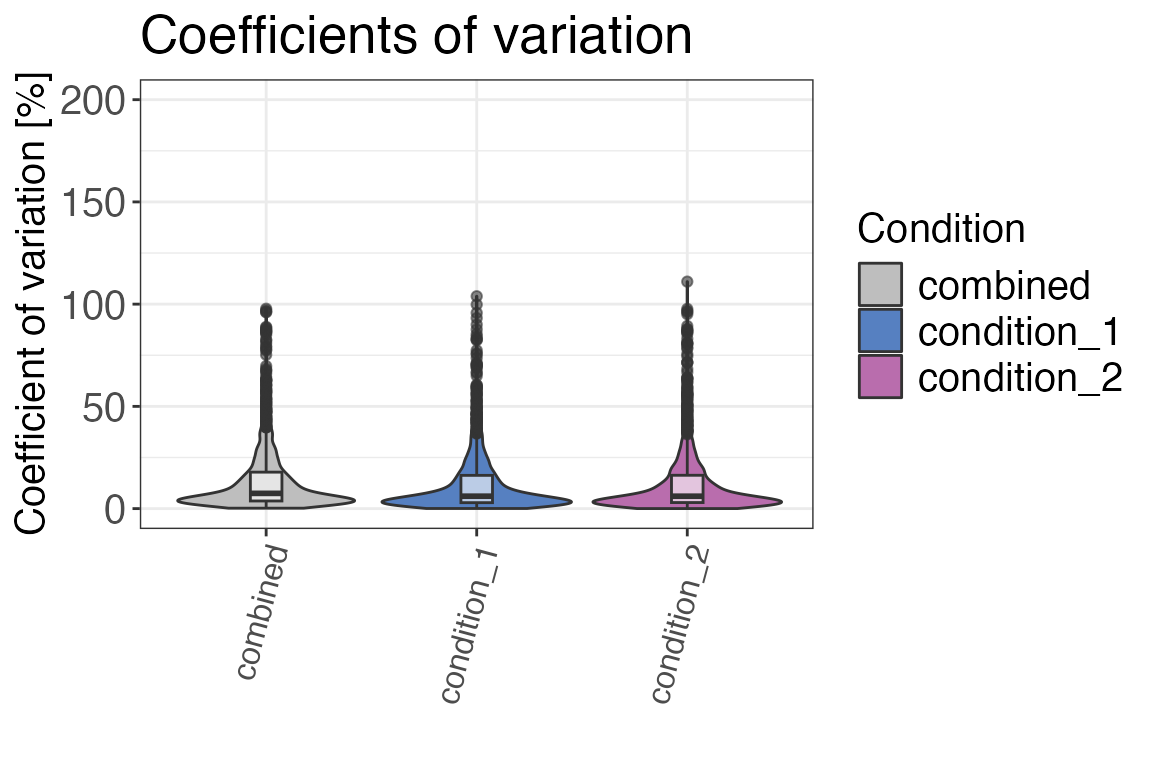
Coefficients of variation
A good first step of quality control is to check if coefficients of
variation between replicates are in a reasonable range. Ideally, these
should be below 15-20 %. If you see groups with higher CVs, there might
have been a sample preparation error or the instrument performance was
not ideal at the time of measurement. You can check if after
normalise() (depending on whether your search engine has
performed normalisation already) your CVs improve.
To conduct the CV-based quality control, we make use of the function
qc_cvs(). This function calculates the coefficients of
variation. The output can either be a table or a plot of coefficients of
variations for each condition. Below we are returning a CV table and a
violin plot, showing the CVs for different conditions.
The CVs (in percent) are calculated with the following formula:
Since the CV function works only with raw values, we will
backtransform our log2 transformed data and create a new column called
raw_intensity from the
peptide_intensity_missing column which contains peptide
intensities. To create the new column, we are using the function
mutate() from the R package dplyr. We also
make use of the pipe operator %>% included in the R
package magrittr which takes the output of the preceding
function and supplies it as the first argument of the following
function. Using %>% makes code easier to read and
follow.
The “combined” group of CVs contains CVs across all samples and not only across the replicates of a certain condition. Ideally the combined CVs should always be the group with the highest CV. This would indicate that CVs within conditions are low and the treatment had and effect that causes an increase in overall CVs. If the treatment did not have an effect or only a very small one then the combined CV could be as high as the CVs of individual conditions. If an individual condition has a higher CV than the combined CV then there was potentially a problem with one or multiple of the samples. You can check if there are suspicious samples behaving differently from the rest in the other quality control metrics. If there is a consistent different behaviour these samples should potentially be excluded for analysis. Sometimes a condition can also just be noisy because of the treatment in this condition that causes a larger variation. Therefore comparing individual CVs with the combined CV should only be done with caution.
input <- data %>%
# as the data is log2 transformed, we need to transform it back before calculating the CVs
mutate(raw_intensity = 2^peptide_intensity_missing)
qc_cvs(
data = input,
grouping = peptide,
condition = condition,
intensity = raw_intensity,
plot = FALSE
)
#> # A tibble: 2 × 3
#> condition median_cv median_cv_combined
#> <chr> <dbl> <dbl>
#> 1 condition_2 6.06 7.49
#> 2 condition_1 6.07 7.49
qc_cvs(
data = input,
grouping = peptide,
condition = condition,
intensity = raw_intensity,
plot = TRUE,
plot_style = "violin"
)
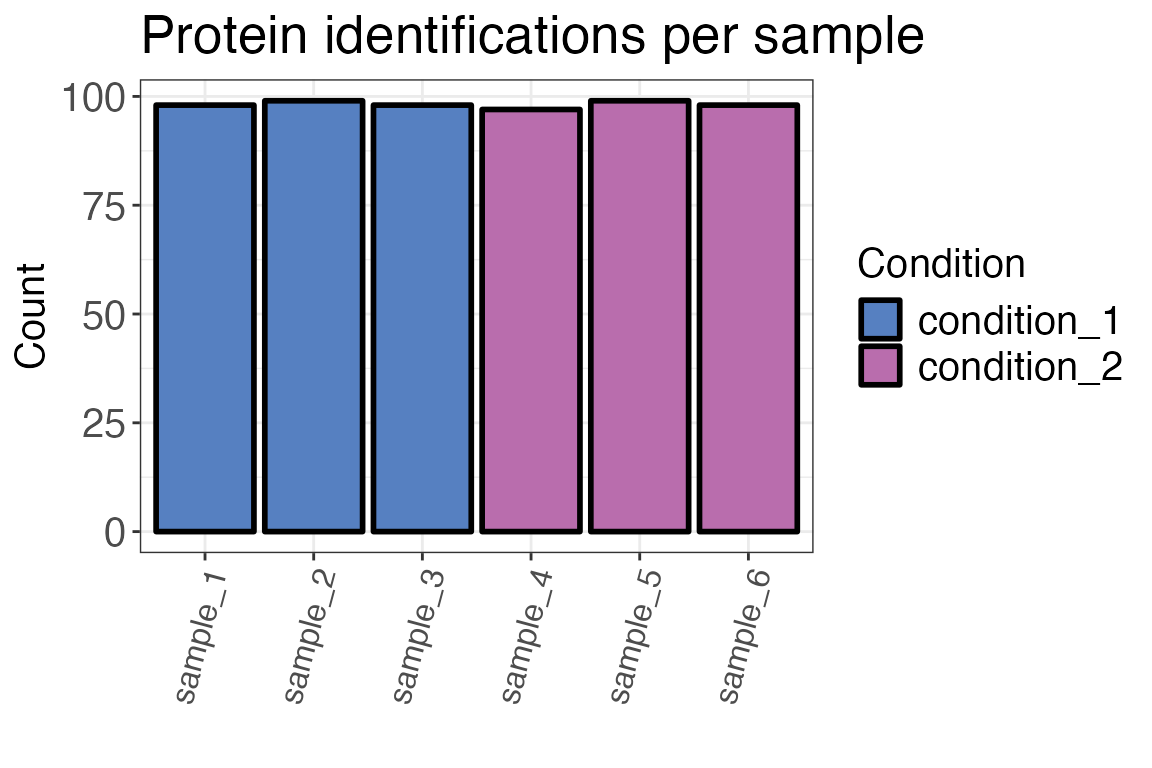
Number of identifications (IDs)
The number of protein or peptide identifications should be similar for different samples. If there are significanlty less observations in one sample, this might indicate a sample preparation or measurement error.
For the analysis of the number of identifications of precursors,
peptides or proteins we use the function qc_ids(). This
function can return either a table or a plot. The output of this
function - and also of a lot of other protti functions
- can be plotted in an interactive plot that makes use of the R package
plotly. You can plot an interactive version of the plot by
setting interactive = TRUE within the function call.
qc_ids(
data = input,
sample = sample,
grouping = protein,
intensity = peptide_intensity_missing,
condition = condition,
plot = FALSE
)
#> # A tibble: 6 × 3
#> sample condition count
#> <fct> <chr> <int>
#> 1 sample_3 condition_1 98
#> 2 sample_4 condition_2 97
#> 3 sample_5 condition_2 99
#> 4 sample_6 condition_2 98
#> 5 sample_2 condition_1 99
#> 6 sample_1 condition_1 98
qc_ids(
data = input,
sample = sample,
grouping = protein,
intensity = peptide_intensity_missing,
condition = condition,
title = "Protein identifications per sample",
plot = TRUE
)
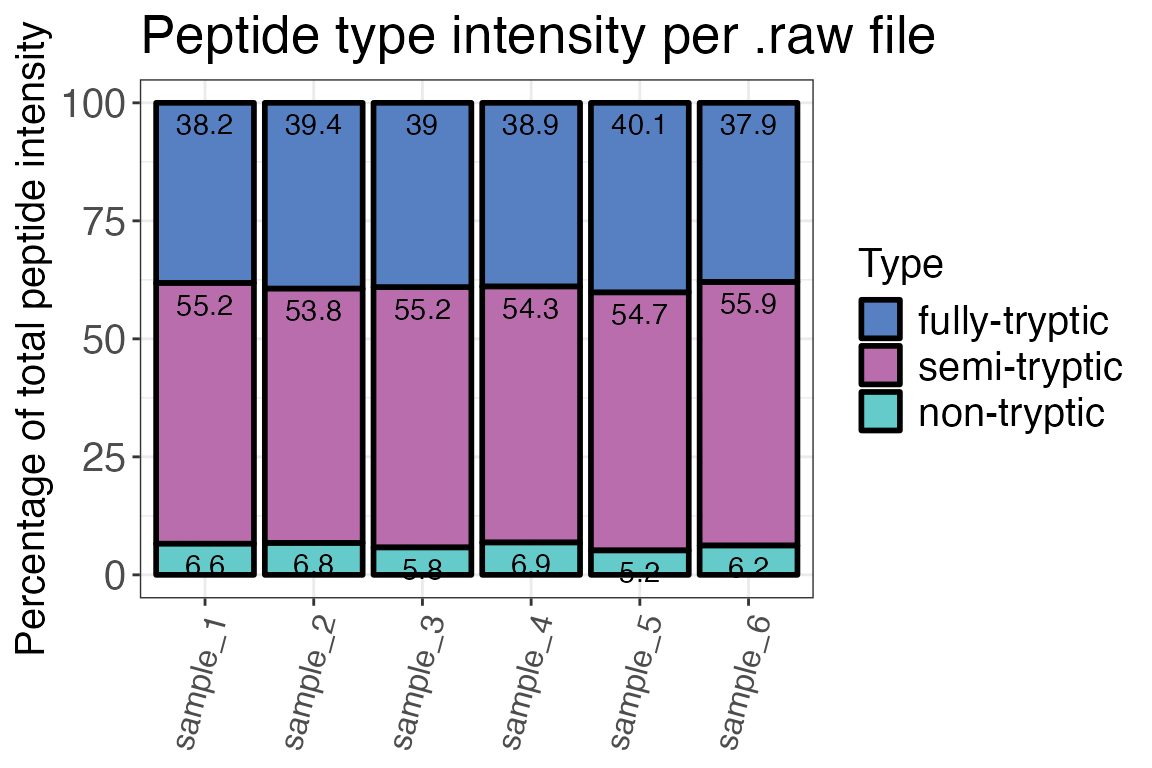
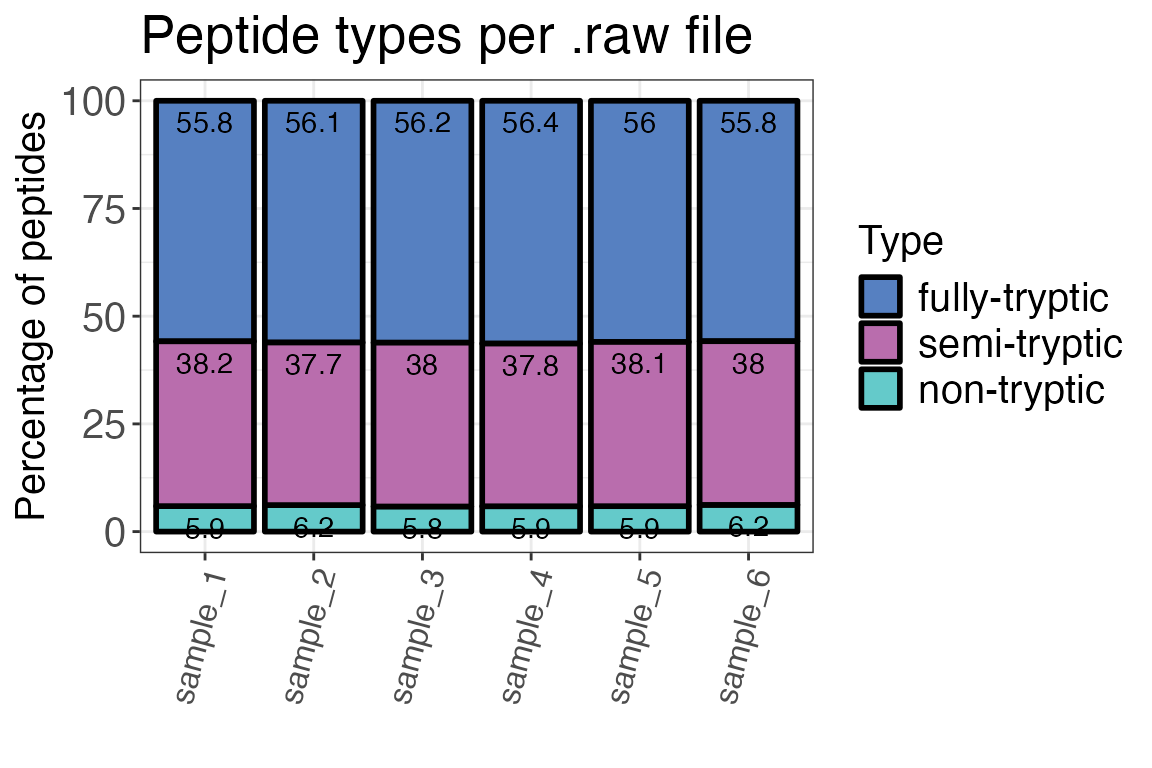
Peptide types
Now that we had a look at protein IDs, we should also check peptide identifications for homogeneity across samples. An important factor to take into account when analysing LiP-MS data are peptide types (tryptic, semi-tryptic, non-tryptic). They can give us an idea about the reproducibility of cleavage events in the experiment and if our treatment influenced protease activity or the digest in general.
In a standard LiP-MS experiment we use 3 different proteases:
- Proteinase K (unspecific, cleaves accessible protein regions)
- Trypsin (specific, cleaves after lysines or arginines)
- Lys-C (specific, cleaves after lysines)
Peptide types can be assessed in Spectronaut directly or by using the
protti function assign_peptide_type().
This function takes amino acids before and after the first and last
amino acid in the peptide into account. A peptide that starts after a
lysine or arginine and ends with a lysine or arginine is a
tryptic peptide. A peptide that has one proteinase K cleavage
site (not lysine or arginine) and one trypsin cleavage site either at
the beginning or at the end of the peptide is a semi-tryptic
peptide. A peptide that does not have any trypsin cleavage sites is a
non-tryptic peptide. If information about the amino acid before
and the last amino acid of a peptide are not available in your data you
can use the find_peptide function from the
protti package to create it.
We are going to use the function qc_peptide_type() to
evaluate the distribution of peptide types between our samples. The
function can return a plot or a table, based on either peptide type
counts or intensities. Both are equally important when assessing how
homogenous the samples are. For method = intensity the
function uses raw intensity values, so we are going to use the column
raw_intensity which we added previously.
In a typical LiP-MS experiment, there should be few non-tryptic peptides. The peptide type distribution for fully- and semi-, and non-tryptic peptides depends on your digestion conditions (i.e. temperature, duration, concentration).
qc_peptide_type(
data = input,
sample = sample,
peptide = peptide,
pep_type = pep_type,
method = "intensity",
intensity = raw_intensity,
plot = TRUE,
interactive = FALSE
)
qc_peptide_type(
data = input,
sample = sample,
peptide = peptide,
pep_type = pep_type,
method = "count",
plot = TRUE,
interactive = FALSE
)
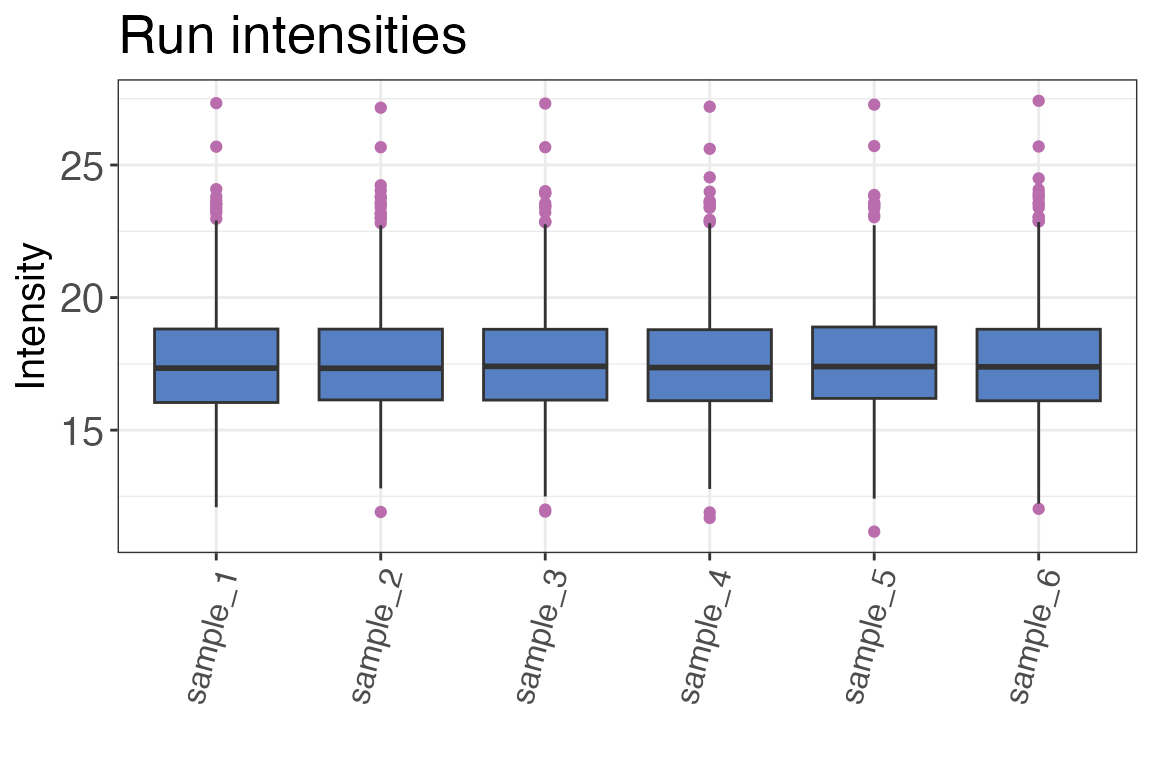
Run intensities
The function qc_intensity_distribution() plots all
precursor, peptide or protein intensities for each sample as a boxplot
if method “boxplot” is selected. This is helpful for quick assessment of
any major sample losses or measurement issues.
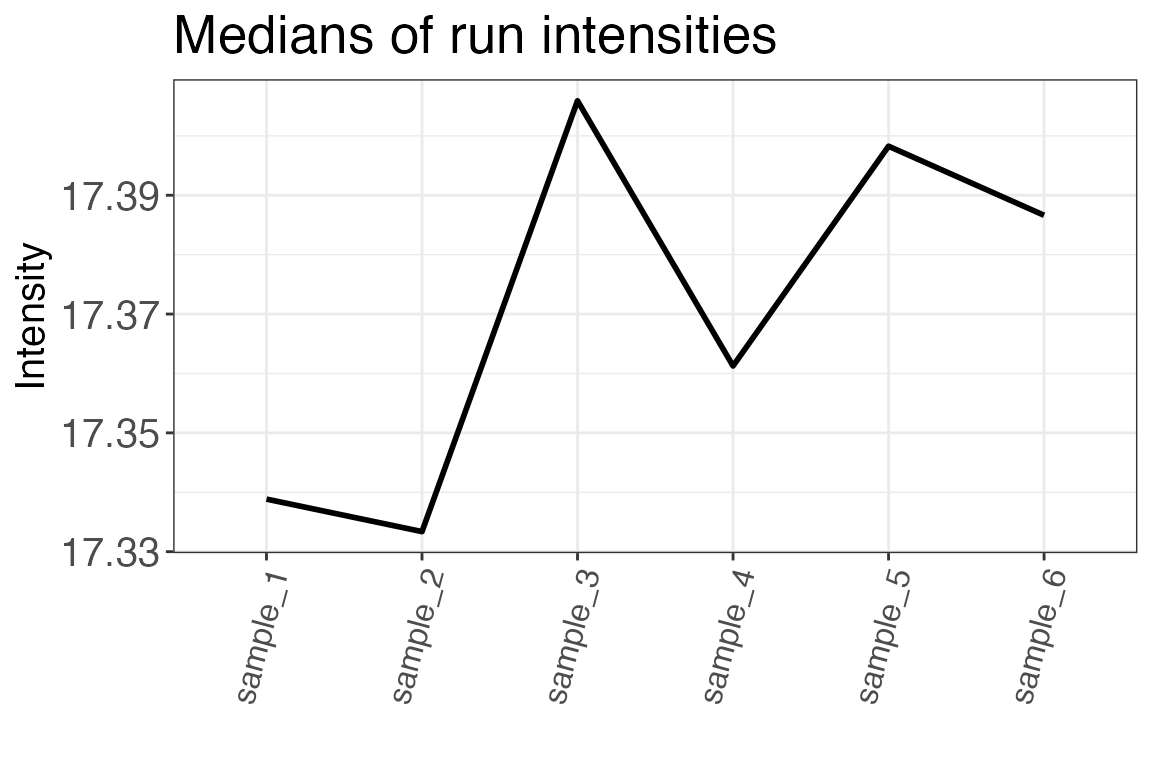
Run intensities can also be assessed by plotting the median run
intensities as a line plot. This helps you quickly assess if there are
any trends in your data. The function to use for this analysis is
qc_median_intensities().
qc_intensity_distribution(
data = input,
sample = sample,
grouping = peptide,
intensity_log2 = peptide_intensity_missing,
plot_style = "boxplot"
)
qc_median_intensities(
data = input,
sample = sample,
grouping = peptide,
intensity = peptide_intensity_missing
)
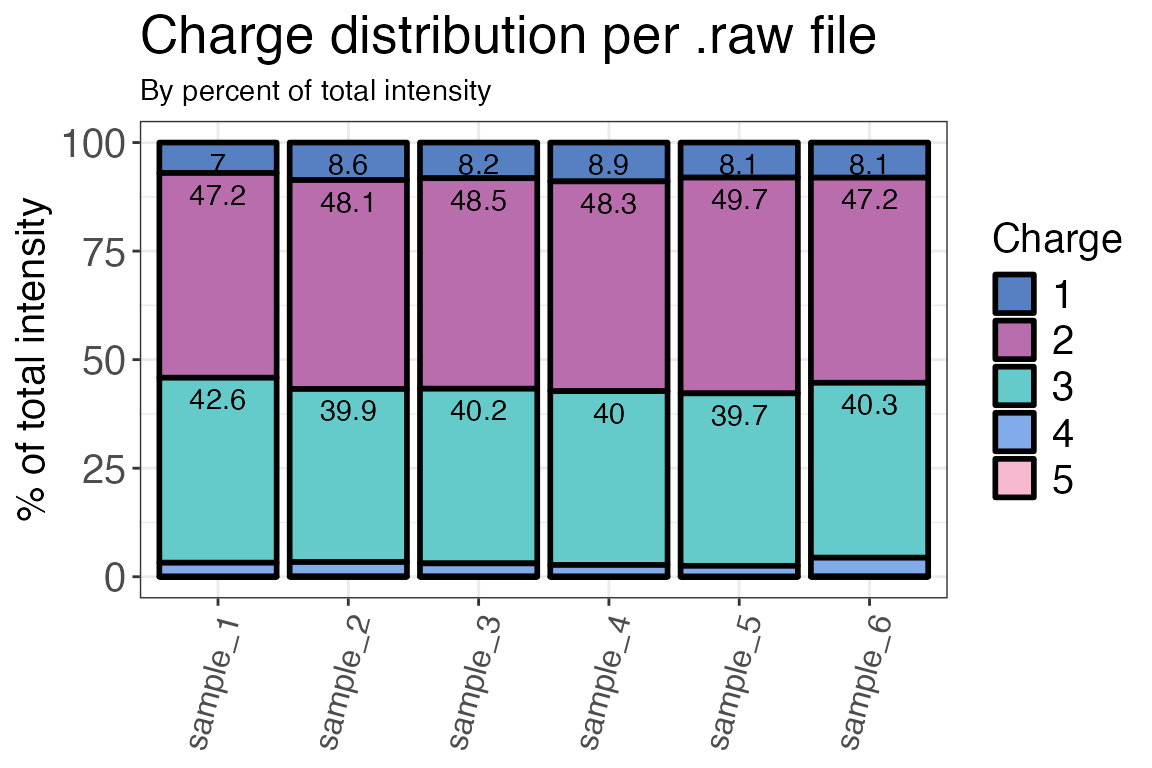
Charge states
The charge state distibution of the detected peptides can be assessed
with qc_charge_states(). Similar to the peptide types, this
should also be homogenous for the different samples. The function can
return either a plot or a table based on counts of peptides with a
specific charge state or the intensities of these peptides. For
method = "intensity" the function requires the raw
intensity values created previously as its input.
qc_charge_states(
data = input,
sample = sample,
grouping = peptide,
charge_states = charge,
method = "intensity",
intensity = raw_intensity,
plot = TRUE
)
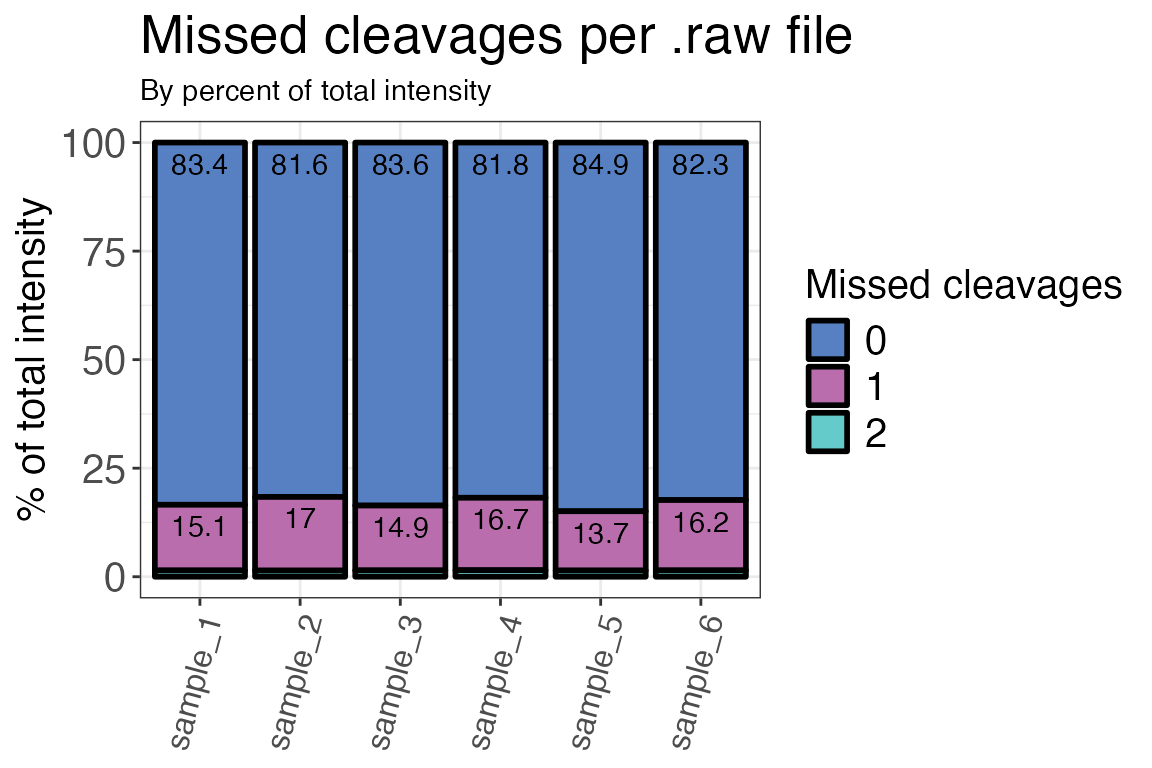
Missed cleavages
An important measure of efficient digests with e.g. trypsin are the number of missed cleavages. The proteases trypsin and Lys-C both cleave after positively charged amino acids lysine (K) and arginine (R). If a measured peptide includes either K or R within its sequence, i.e. not at the last position, it contains a missed cleavage site. These peptides are important to take into account because they can tell you if your tryptic digest was inefficient. The number of missed cleavages should generally be low in a proteomics or LiP-MS dataset.
We are going to check the numbers of missed cleavages in our dataset
by using the function qc_missed_cleavages(). It can assess
missed cleavages based on the count of peptides with missed cleavages or
the intensities of the corresponding peptides. For
method = "intensity" the function uses the raw (not log2
transformed) intensity values. You can have the function either return a
plot or a table.
qc_missed_cleavages(
data = input,
sample = sample,
grouping = peptide,
missed_cleavages = n_missed_cleavage,
method = "intensity",
intensity = raw_intensity,
plot = TRUE
)
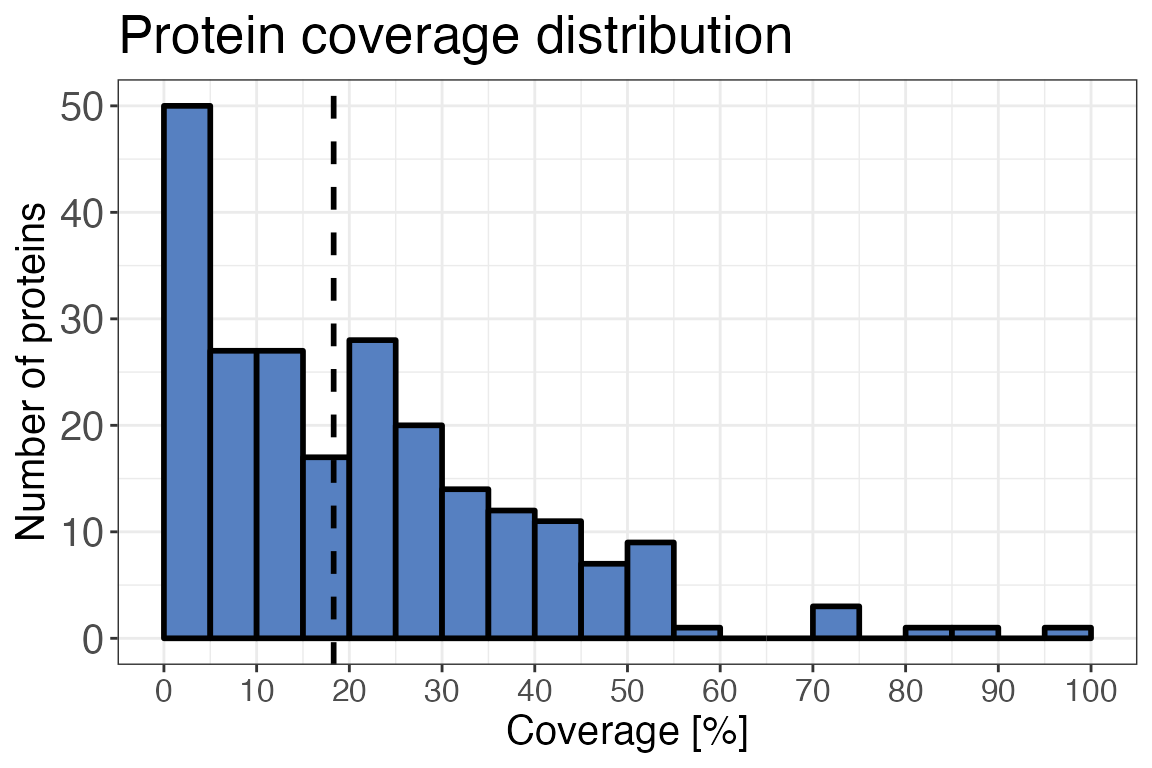
Sequence coverage
The following function gives you insight into the protein coverage (i.e. what percentage of the protein sequence is covered by the identified peptides) in the dataset. For a LiP-MS experiment, or any other peptide-centric proteomics experiment, high sequence coverages are desirable.
To assess the protein coverage distribution, we are going to use the
function qc_protein_coverage(). If you do not have a column
containing the protein coverages in your data, you can use the function
calculate_sequence_coverage() to obtain this
information.
qc_sequence_coverage(
data = input,
protein_identifier = protein,
coverage = coverage
)
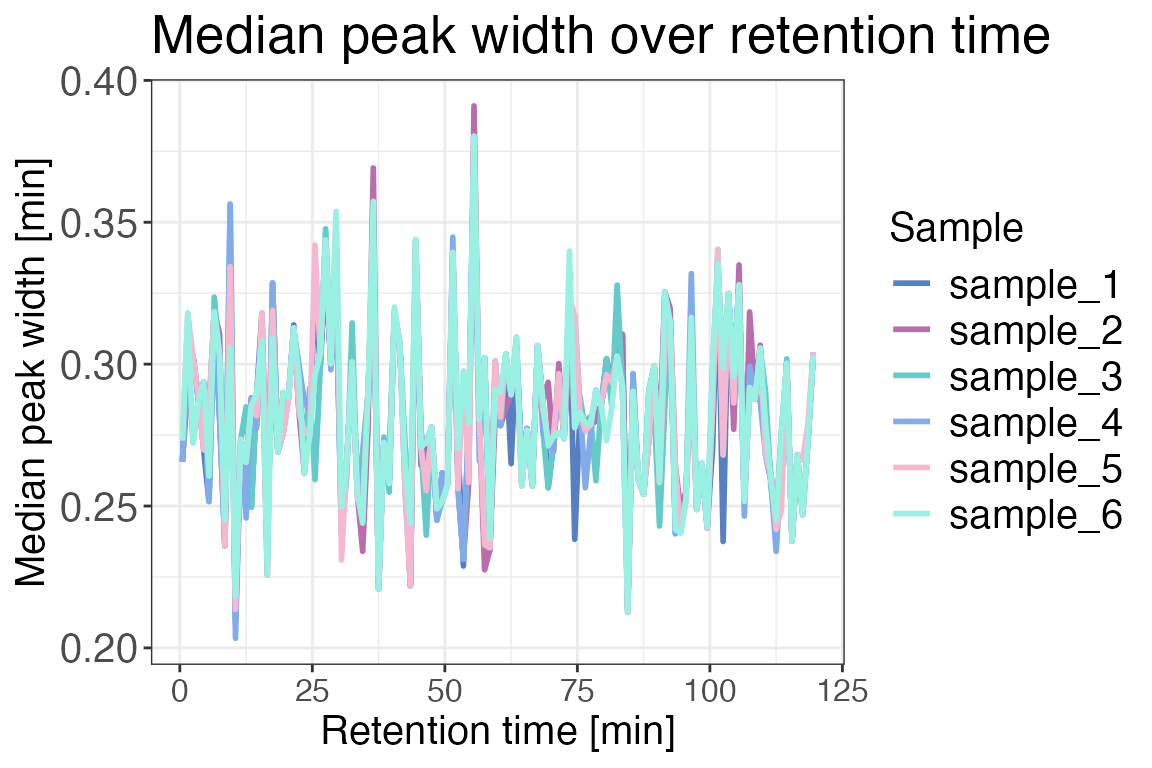
Peak width
In order to identify potential chromatographic issues that might have
occurred during the measurement we can have a look at the
chromatographic median peak widths over the complete chromatogram. To do
this we are going to use the function qc_peak_width(). This
function requires either the peak start and end times or the retention
time and peak width. This information can be obtained from Spectronaut
(or any other search engine of your choice). The peak widths should be
similar for all the measured samples of the experiment.
qc_peak_width(
data = input,
sample = sample,
intensity = peptide_intensity_missing,
retention_time = retention_time,
peak_width = peak_width
)
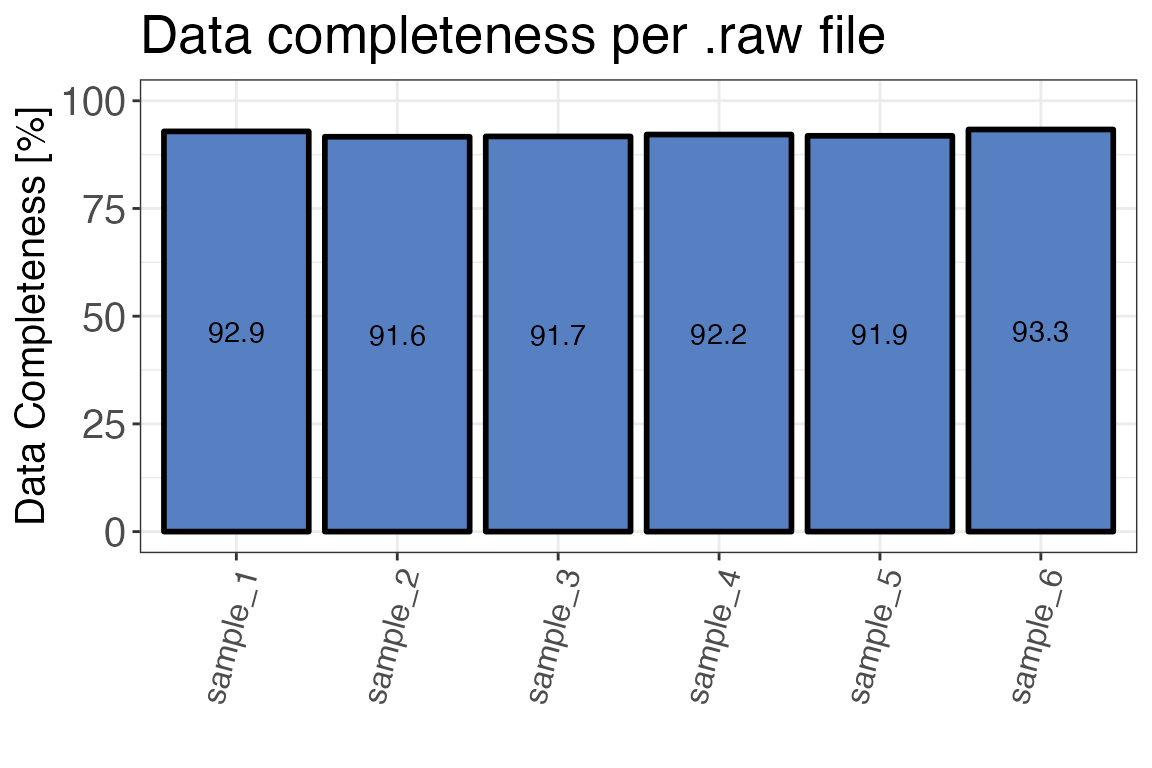
Data completeness
The function qc_data_completeness() checks how many of
all detected precursors, peptides or proteins were identified in each
sample. The function can return either a plot or a table.
qc_data_completeness(
data = input,
sample = sample,
grouping = peptide,
intensity = peptide_intensity_missing,
plot = TRUE
)
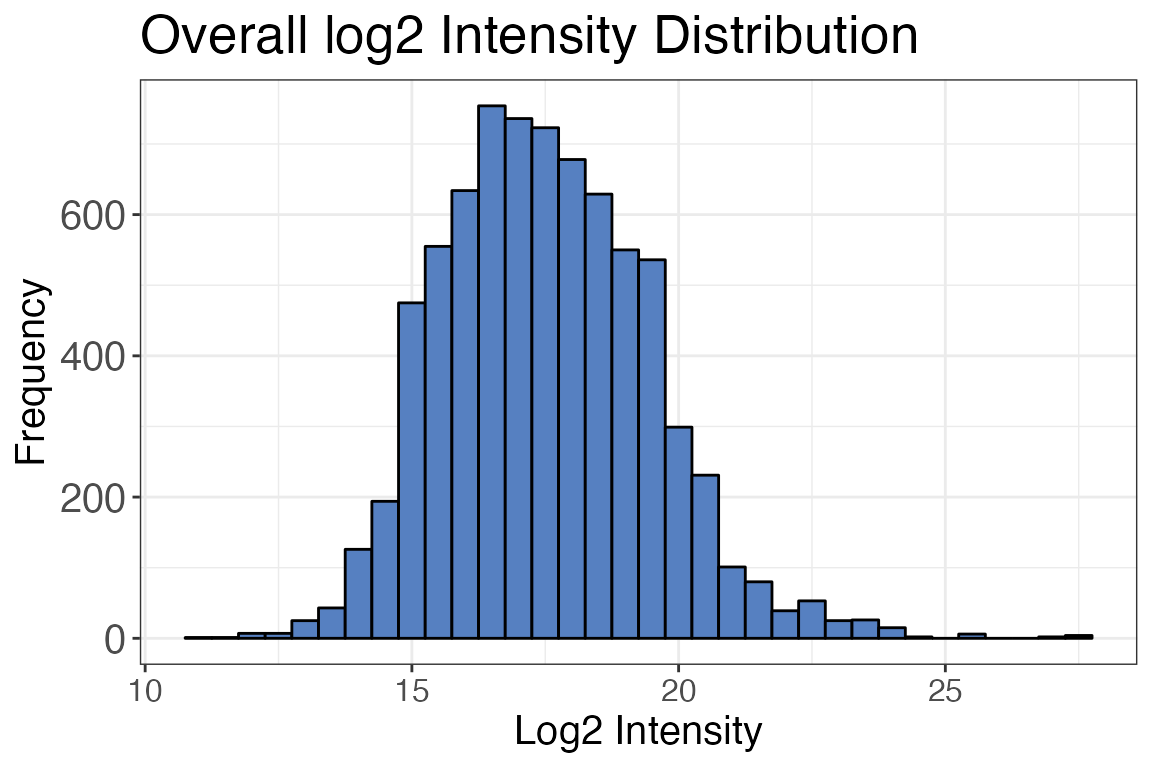
Log2 Intensity distribution
For different kinds of analyses (e.g. t-tests) it is important that
your data intensity follows a normal distribution. To ensure that this
is the case, we are going to use the function
qc_intensity_distriubution(). The function returns a
histogram plot when plot_style = "histogram" showing how
the intensities are distributed.
qc_intensity_distribution(
data = input,
grouping = peptide,
intensity_log2 = peptide_intensity_missing,
plot_style = "histogram"
)
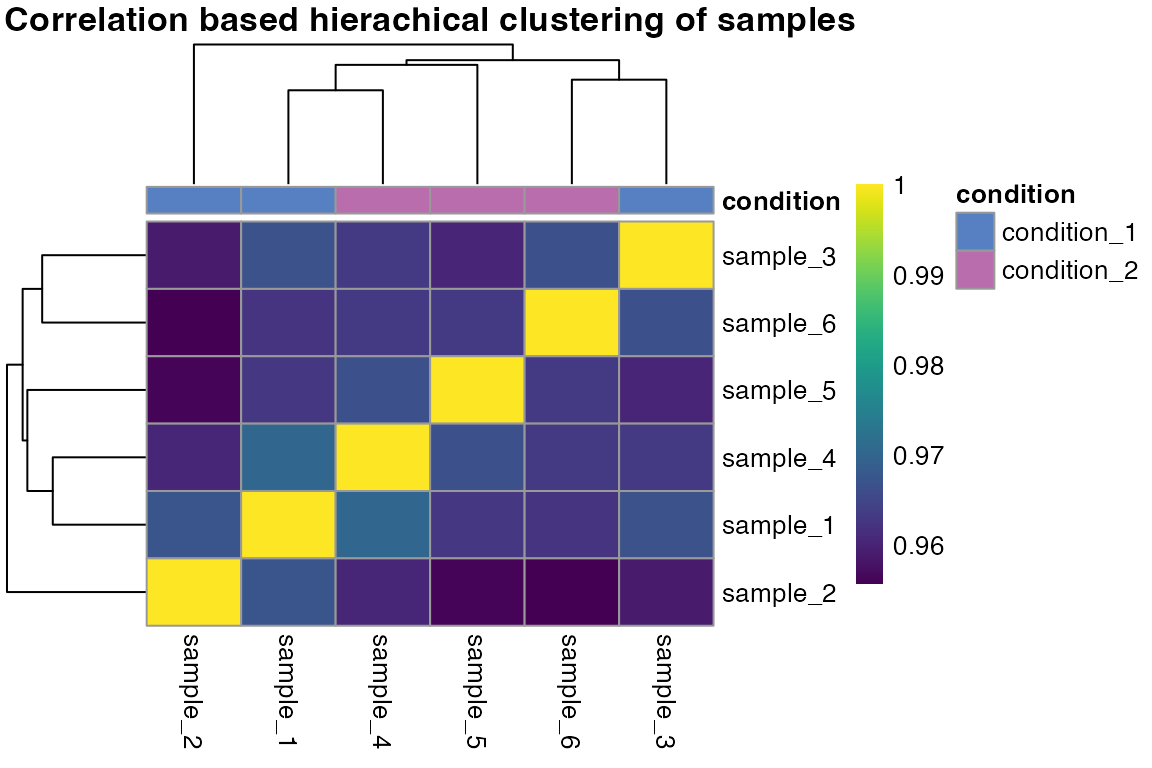
Sample correlation
Another approach to quality control is to check the correlation of
your samples. Ideally, replicates should cluster together and different
treatment conditions should be separated. We are now going to check if
this is the case for our data by using the function
qc_sample_correlation(). The function will return a
correlation heatmap with a comparison of all samples.
qc_sample_correlation(
data = input,
sample = sample,
grouping = peptide,
intensity_log2 = peptide_intensity_missing,
condition = condition,
interactive = FALSE
)
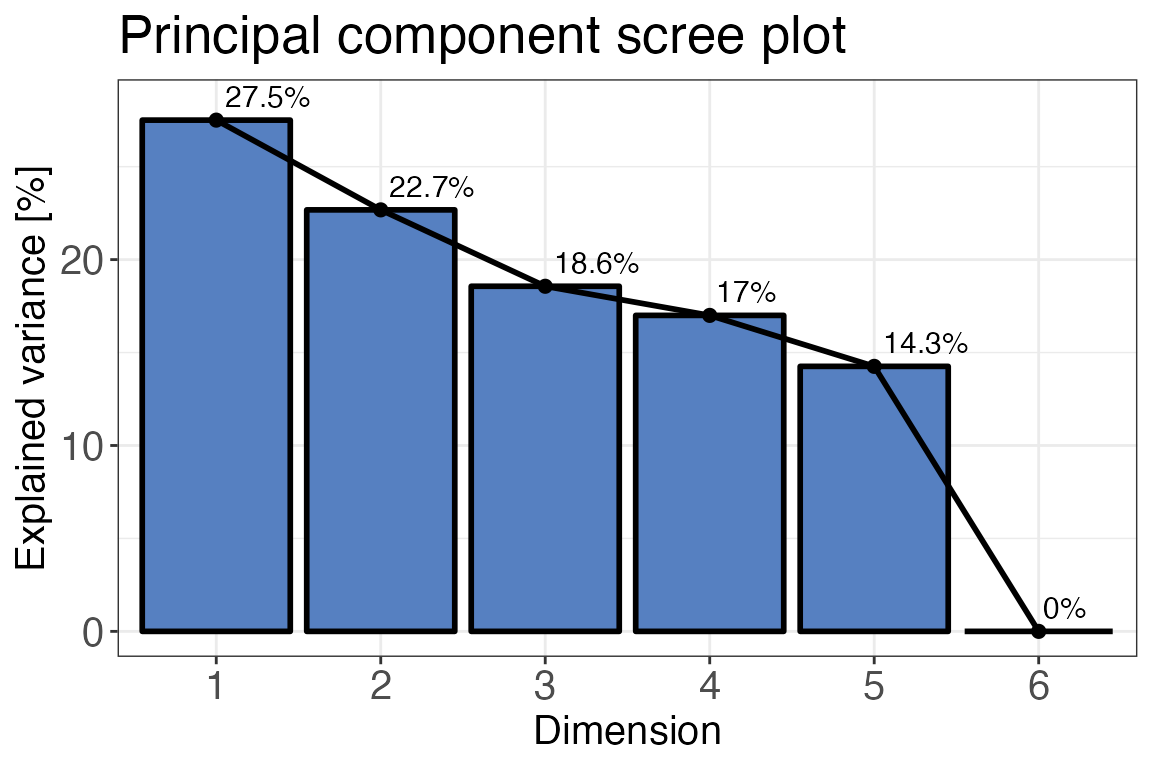
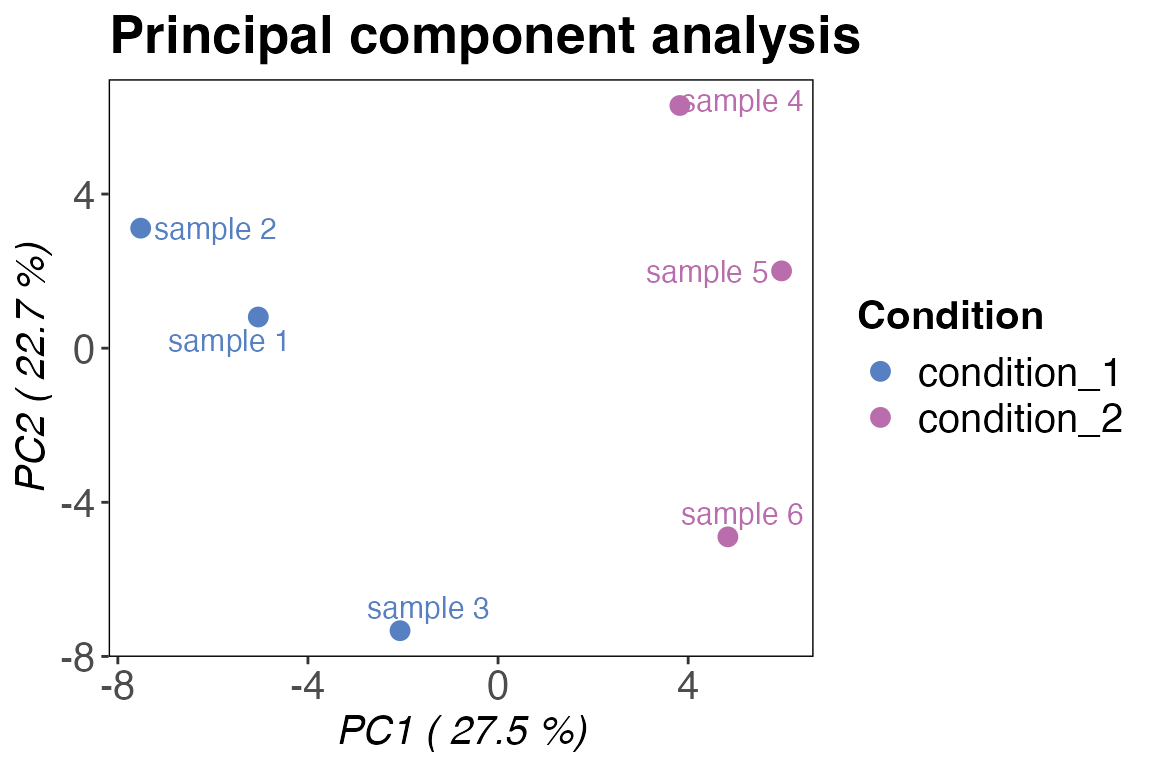
Principal component analysis (PCA)
Another popular quality control method (which could also be
considered part of data analysis) is the principal component analysis
(PCA). Generally, PCA is a method that reduces dimensionality of large
datasets. In our case this helps us to quickly assess how similar or
different our replicates and conditions are. We are going to use
qc_pca() to compute and plot a PCA for our data. Before
plotting your PCA you can check how much of your variance can be
explained with each of the principle components. To do that you can plot
a scree-plot using the function qc_pca() and setting
plot_style = "scree". In this example we can see that the
variance can be best explained with PC1 and PC2 so we can go ahead and
plot these.
qc_pca(
data = data,
sample = sample,
grouping = peptide,
intensity = peptide_intensity_missing,
condition = condition,
digestion = NULL,
plot_style = "scree"
)
qc_pca(
data = data,
sample = sample,
grouping = peptide,
intensity = peptide_intensity_missing,
condition = condition,
components = c("PC1", "PC2"),
plot_style = "pca"
)
Ranked intensity distribution
It can be useful to check which precursors, peptides or proteins are
the most or least abundant in the sample by ranking their intensities.
You can create a data frame containing ranked median intensities for the
whole experiment or ranked intensities for each sample using the
qc_ranked_intensities() function. In addition you can also
create a corresponding plot by setting the plot argument to
TRUE. If you would like to axis to not be log10 transformed
but rather log2 you can change the y_axis_transformation
argument.
# Plot ranked peptide intensities
qc_ranked_intensities(
data = data,
sample = sample,
grouping = peptide,
intensity_log2 = peptide_intensity,
plot = TRUE,
)
#> Warning: ggrepel: 17 unlabeled data points (too many overlaps). Consider
#> increasing max.overlaps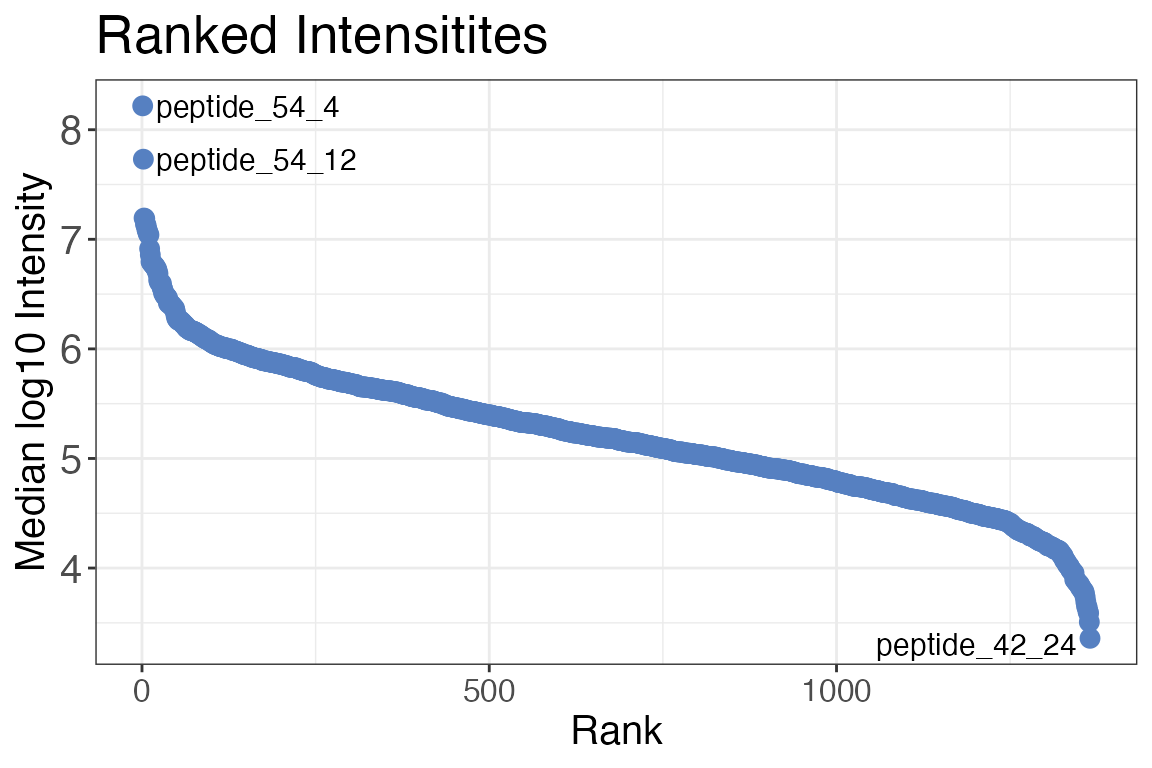
Additional QC functions
There are two additional QC functions that are not included in this vignette:
-
qc_contaminants()is a function that can be used if proteins are assigned as contaminants. This information can for example be exported from MaxQuant. The function calculates the percentage of proteins annotated to be contaminants and returns either a table or a plot. -
qc_proteome_coverage()calculates the proteome coverage for each sample and the whole experiment and returns either a plot or a table.