Single Dose Treatment Data Analysis Workflow
Dina Schuster
2026-01-14
Source:vignettes/data_analysis_single_dose_treatment_workflow.Rmd
data_analysis_single_dose_treatment_workflow.RmdIntroduction
This vignette will give you an overview of how you can analyse bottom-up proteomics data or LiP-MS data using protti. If you would like to analyse dose-response data please refer to the dose-response data analysis vignette. Before analysing your data make sure that it is of sufficient quality and that you do not have any outliers. To do this you can take a look at the quality control vignette.
protti includes several functions that make it easy for the user to analyse and interpret data from bottom-up proteomics or LiP-MS experiments. The R package includes functions for
- Quality control (see quality control vignette for more detailed information)
- Data preparation
- Median normalisation
- Data filtering
- Protein abundance calculation from precursor or peptide intensities
- Imputation of missing values
- Fetching of database information (ChEBI, GO, KEGG, MobiDB, UniProt)
- Calculation of sequence coverage
- Data analysis
- Statistical hypothesis tests
- Fitting of dose-response curves (relevant for experiments with several treatment concentrations, click here for more information)
- Data visualisation
- Volcano plots
- Barcode plots (plots that show protein coverage and significant changes projected onto the protein sequence)
- Wood’s plots
- Profile plots
- Data interpretation
- Treatment enrichment (check if your hits are enriched with known targets)
- GO-term enrichment
- Network analysis (based on STRING database information)
- KEGG pathway enrichment
You can read more about specific functions and how to use them by
calling e.g. ?normalise (for the normalise()
function). Calling ? followed by the function name will
display the function documentation and give you more detailed
information about the function. This can be done for any of the
functions included in the package.
This document will give you an overview of data preparation, data analysis, data visualisation and data interpretation functions included in protti. It will show you how they can be applied to your data. The examples in this file are run on a published LiP-MS data set. In the experiment a HeLa cell lysate was treated with different amounts of the drug rapamycin and the target of rapamycin (FKBP12) was successfully identified. For this vignette we are using a filtered version of the original data set where we include only the 10 μM treatment concentration and the control (untreated). For simplicity only 50 proteins are included in the data set.
The data set is produced from the output of Spectronaut™.
However, if you have any other data such as DDA data that was searched
with a different search engine you can still apply
protti’s functions. Just make sure that your data frame
contains tidy data.
That means data should be contained in a long format (e.g. all sample
names in one column) rather than a wide format (e.g. each sample name in
its own column). You can easily achieve this by using the
pivot_longer() function from the tidyr
package. If you are unsure what your input data is supposed to look
like, please use the create_synthetic_data() function and
compare this to your data. You can also take a look at the input
preparation vignette, there you will find all the necessary
information on how to get your data into the correct format.
The input data should have a similar structure to this example:
| Sample | Replicate | Peptide | Condition | log2(Intensity) |
|---|---|---|---|---|
| sample1 | 1 | PEPTIDER | treated | 14 |
| sample1 | 1 | PEPTI | treated | 16 |
| sample1 | 1 | PEPTIDE | treated | 17 |
| sample2 | 1 | PEPTIDER | untreated | 15 |
| sample2 | 1 | PEPTI | untreated | 18 |
| sample2 | 1 | PEPTIDE | untreated | 12 |
How to use protti to analyse your data
Getting started
Before we can start analysing our data, we need to load the
protti package. This is done by using the base R
function library(). In addition, we are also loading the
packages magrittr and dplyr. Both
magrittr and dplyr are part of the tidyverse, a collection of packages
that provide useful functionalities for data processing and
visualisation. If you use many tidyverse packages in your workflow you
can easily load all at once by calling
library(tidyverse).
After having loaded the required packages we will load our data set
into the R environment. In order to do this for your data set you can
use the function read_protti(). This function is a wrapper
around the fast fread() function from the data.table
package and the clean_names() function from the janitor
package. This will allow you to not only load your data into R very
fast, but also to clean up the column names into lower snake case to
make them more R-friendly. This will make it easier to remember them and
to use them in your data analysis.
# To read in your own data you can use read_protti()
your_data <- read_protti(filename = "mydata/data.csv")For this example we are going to use the rapamycin_10uM
test data set included in protti. To read in the file
we are simply going to use the utils function
data().
data("rapamycin_10uM")Data preparation
Log2 transformation, median normalisation and CV filtering
After inspecting the data and performing quality control (see quality control vignette for more information) we will now start to prepare the data for the analysis.
First, we remove decoy hits (used for false discovery rate
estimation). Our example data set contains a column called
eg_is_decoy that consists of logicals indicating whether or
not the peptide is a decoy hit. To remove decoys we will use the
dplyr function filter().
Next, we log2 transform our intensities, then normalise the data to
the median value of all runs. To transform the intensities we use the
dplyr function mutate() which creates a new
column while maintaining the original column.
Note that we are also using the pipe operator %>%
included in the R package magrittr.
%>% takes the output of the preceding function and
supplies it as the first argument of the following function. Using
%>% makes code easier to read and follow.
For normalisation we are using the protti function
normalise(). For this example we will use median
normalisation (method = "median"). The function normalises
intensities for each run to the median of all runs. This is only
necessary if your search algorithm does not already median normalise
your intensities. For the example data we have disabled median
normalisation in Spectronaut, therefore we need to median normalise now.
The formula for median normalisation is:
To ensure that only good peptide measurements will be used for
further analysis, we will also filter our data based on coefficients of
variation (CV). In order to do this we are using the function
filter_cv(). For this example we are retaining peptides
with a CV < 25 % in at least one of the two conditions.
The CVs are calculated within the function according to the formula:
Note: The use of the filter_cv() function is
optional. It might remove a lot of your data if your experiment was
noisy. However, especially in these cases, the function will remove
peptides with poor quality and should improve the result. It is very
important that if you use this function you should not use the moderated
t-test or proDA algorithm on your data for differential abundance
estimation and significance testing. This likely will lead to an
inflated false positive rate because it alters the distributional
assumptions of these tests (Bourgon
2010).
data_normalised <- rapamycin_10uM %>%
filter(eg_is_decoy == FALSE) %>%
mutate(intensity_log2 = log2(fg_quantity)) %>%
normalise(
sample = r_file_name,
intensity_log2 = intensity_log2,
method = "median"
)
data_filtered <- data_normalised %>%
filter_cv(
grouping = eg_precursor_id,
condition = r_condition,
log2_intensity = intensity_log2,
cv_limit = 0.25,
min_conditions = 1
)Remove non-proteotypic peptides
For LiP-MS analysis we commonly remove non-proteotypic peptides (i.e. peptides that could come from more than one protein). If you detect a change in non-proteotypic peptides it is not possible to clearly assign which protein it comes from and therefore which protein is affected by your treatment. If you are using the output from Spectronaut you will find a column called “pep_is_proteotypic”. This column contains logicals indicating whether your peptide is proteotypic or not.
To filter out non proteotypic peptides we are using the
dplyr function filter().
Fetching database information and assigning peptide types
In order to obtain more information about our identified proteins we
are going to use the function fetch_uniprot() to download
information from UniProt directly.
fetch_uniprot() uses a vector of UniProt IDs as its
input. We produce this vector by using the base R function
unique() which will extract all unique elements in the
selected protein ID column. In this case we want to download the full
protein name, gene IDs, GO terms associated with molecular function,
StringDB IDs, information on known interacting proteins, location of the
active site, location of binding sites, PDB entries, protein length and
protein sequence. There are more options for columns to add (for more
information on possible columns to add click here).
fetch_uniprot() returns a new data frame. In order to be
able to merge this with our original data frame we have to rename the ID
column to match the name of the protein ID column of our original data
frame. To do this we use dplyr’s rename()
function.
To merge the two data frames we use the dplyr function
left_join(). We match the two data frames by the column
“pg_protein_accessions”. By using left_join() we retain all
rows from our original data frame while adding the columns from the data
fame generated with fetch_uniprot().
Note: you can also directly join the UniProt data frame with your
data without the need to rename its id column. You can
specify in the by argument in left_join() that
two columns are differently named.
Next, we would like to assign the trypticity of our peptides (i.e. if
the peptides are fully-tryptic, semi-tryptic or non-tryptic). In order
to do this we first need to define the peptide positions in the protein
and find the preceding and following amino acids. To obtain this
information we use the protti function
find_peptide(). The output of this function can then be
used in the function assign_peptide_type() which will add
an additional column with the peptide trypticity information. By using
the function calculate_sequence_coverage() we add an
additional column to the data frame containing information on how much
of the protein sequence we identified in our experiment.
uniprot_ids <- unique(data_filtered_proteotypic$pg_protein_accessions)
uniprot <-
fetch_uniprot(
uniprot_ids = uniprot_ids,
columns = c(
"protein_name",
"gene_names",
"go_f",
"xref_string",
"cc_interaction",
"ft_act_site",
"ft_binding",
"xref_pdb",
"length",
"sequence"
)
) %>%
rename(pg_protein_accessions = accession)
data_filtered_uniprot <- data_filtered_proteotypic %>%
left_join(
y = uniprot,
by = "pg_protein_accessions"
) %>%
find_peptide(
protein_sequence = sequence,
peptide_sequence = pep_stripped_sequence
) %>%
assign_peptide_type(
aa_before = aa_before,
last_aa = last_aa,
aa_after = aa_after,
protein_id = pg_protein_accessions
) %>%
calculate_sequence_coverage(
protein_sequence = sequence,
peptides = pep_stripped_sequence
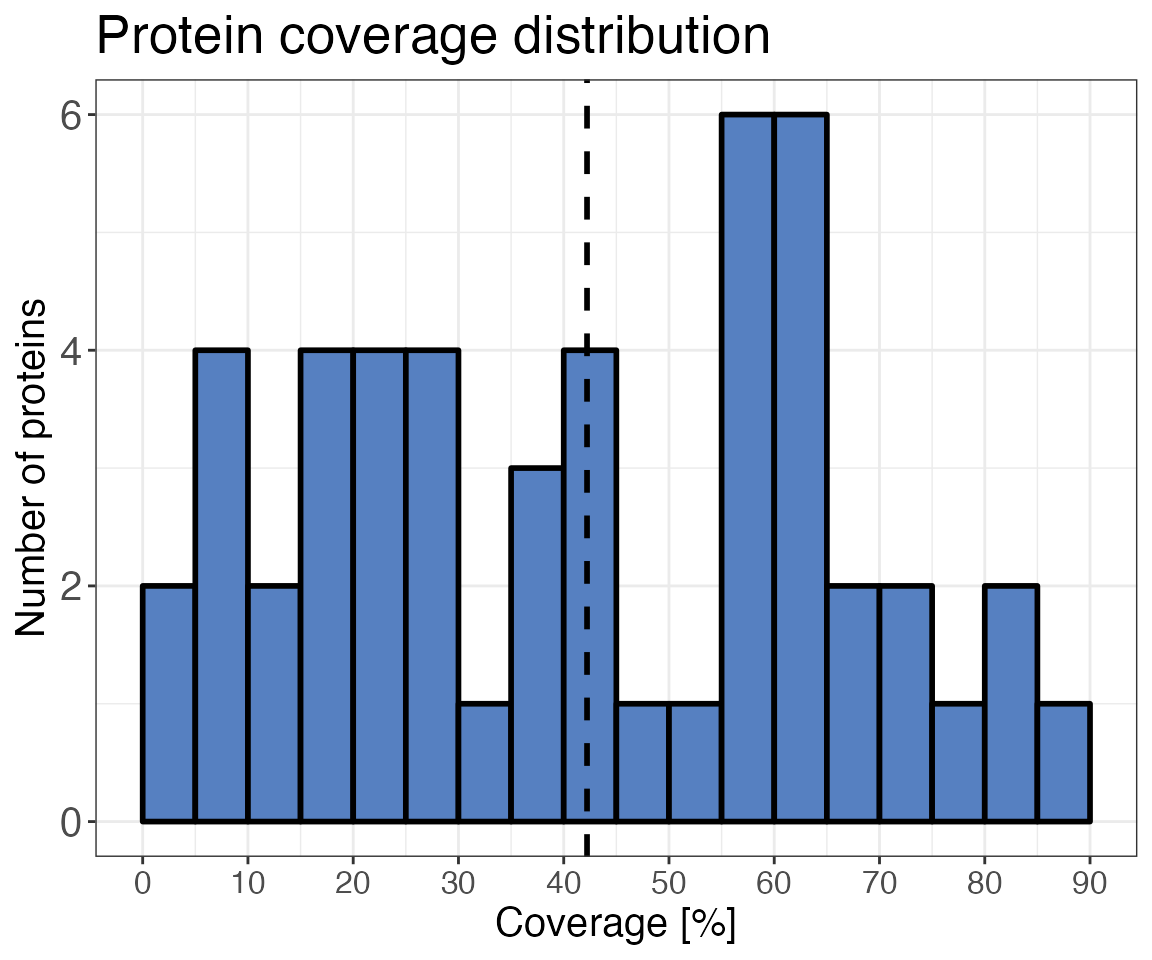
)With the qc_sequence_coverage() function, you check how
sequence coverage is distributed over all proteins in the sample.
Usually, the center of the distribution is low due to many proteins with
poor coverage. For this small data set with only 40 proteins the
sequence coverage is distributed relatively evenly.
qc_sequence_coverage(
data = data_filtered_uniprot,
protein_identifier = pg_protein_accessions,
coverage = coverage
)
Data analysis
Statistical hypothesis test
To test if there is a difference between the peptide abundances in
our two conditions (i.e. rapamycin treated and untreated) we use a
moderated t-test based on the limma R package.
Before the statistical hypothesis test we have to define the types of
missing values present in our data set. We are going to use the function
assign_missingness() which will return a column with
information on the types of missingness we have in our data
(i.e. complete, missing at random (MAR) or missing not at random
(MNAR)). We use the default parameters of this function which assumes
that missing values are MAR when the conditions are at least 70 %
complete (adjusted downward). Missing values are assumed to be MNAR when
less than 20 % of values are present (adjusted_downward) in one
condition if the other condition is complete. If not “complete” all
other comparisons are label as NA. If imputation is
performed, these are the comparisons that will not be imputed. The type
of missingness assigned to a comparison does not have any influence on
the statistical test. However, by default (can be changed) comparisons
with missingness NA are filtered out prior to p-value
adjustment. This means that in addition to imputation the user can use
missingness cutoffs also in order to define which comparisons are too
incomplete to be trustworthy even if significant.
After assigning the types of data missingness we use the function
calculate_diff_abundance(). By selecting
method = t-test the function will perform a Welch’s t-test.
There are also options included to perform a moderated t-test based on
the R package limma or to detect differential abundances
based on the algorithm implemented in the R package proDA.
The algorithm used for proDA is based on a probabilistic
dropout model which facilitates hypothesis testing (using a moderated
t-test) while eliminating the need for imputation.
It has been shown that generally moderated t-tests perform much better also in proteomics data, as compared to t-tests (Kammers et al. 2015). Therefore, we will use a moderated t-test in this example.
Please note that in this example we are not imputing missing values.
You can, however, do so by using the function impute().
This function uses the output of assign_missingness() as
its input. You can use two different imputation methods:
-
method = ludovicwill sample values that are MNAR from a normal distribution around a value that is 3 (log2) lower than the mean intensity of the non-missing condition. The method is was developed by our colleague Ludovic Gillet. -
method = noisewill sample MNAR values from a normal distribution around the mean noise of the complete condition. This requires you to have an additional column with information on the noise, which can be obtained from Spectronaut.
Both methods impute MAR data using the mean and variance of the
condition with the missing data. Missingness assigned as NA
will not be imputed.
Note: If data is imputed this can lead to invalid inferential conclusions due to underestimating statistical uncertainty or it can cause loss of statistical power (Ahlmann-Eltze et al. 2020). Therefore, we do not recommend using a moderated t-test or the proDA algorithm after imputation.
Since we are dealing with a LiP-MS data set we perform the
statistical analysis on the precursor* level. For protein abundance data
you can simply use protein abundances as your intensities and select
your protein groups column for the grouping argument. Make sure to
retain any columns you need for further data analysis with the
retain_columns argument of both functions.
_*A peptide precursor is the actual molecular unit that was detected on the mass spectrometer. This is a peptide with one specific charge state and its modification(s)._
Note: Although it is not required for the data set analysed in this vignette, analysis of LiP-MS data frequently requires correction of LiP peptide intensities for changes in protein abundance. This can be done using the steps outlined in Schopper et al. 2017.
diff_abundance_data <- data_filtered_uniprot %>%
assign_missingness(
sample = r_file_name,
condition = r_condition,
grouping = eg_precursor_id,
intensity = normalised_intensity_log2,
ref_condition = "control",
completeness_MAR = 0.7,
completeness_MNAR = 0.25,
retain_columns = c(
pg_protein_accessions,
go_f,
xref_string,
start,
end,
length,
coverage
)
) %>%
calculate_diff_abundance(
sample = r_file_name,
condition = r_condition,
grouping = eg_precursor_id,
intensity_log2 = normalised_intensity_log2,
missingness = missingness,
comparison = comparison,
method = "moderated_t-test",
retain_columns = c(
pg_protein_accessions,
go_f,
xref_string,
start,
end,
length,
coverage
)
)p-value distribution
The p-value calculated with the moderated t-test is automatically
adjusted for multiple testing using the Benjamini-Hochberg correction.
This assures that we keep the false discovery rate low. An assumption of
this correction is however, that p-values should have an overall uniform
distribution. If there is an effect in the data, there will be an
increased frequency of low p-values. You can check this by using the
protti function pval_distribution_plot(). This also helps
you assess whether your p-value distribution fulfills the assumptions
for your selected FDR control. The cp4p R package is
another great way to check the assumptions underlying FDR control in
quantitative experiments.
pval_distribution_plot(
data = diff_abundance_data,
grouping = eg_precursor_id,
pval = pval
)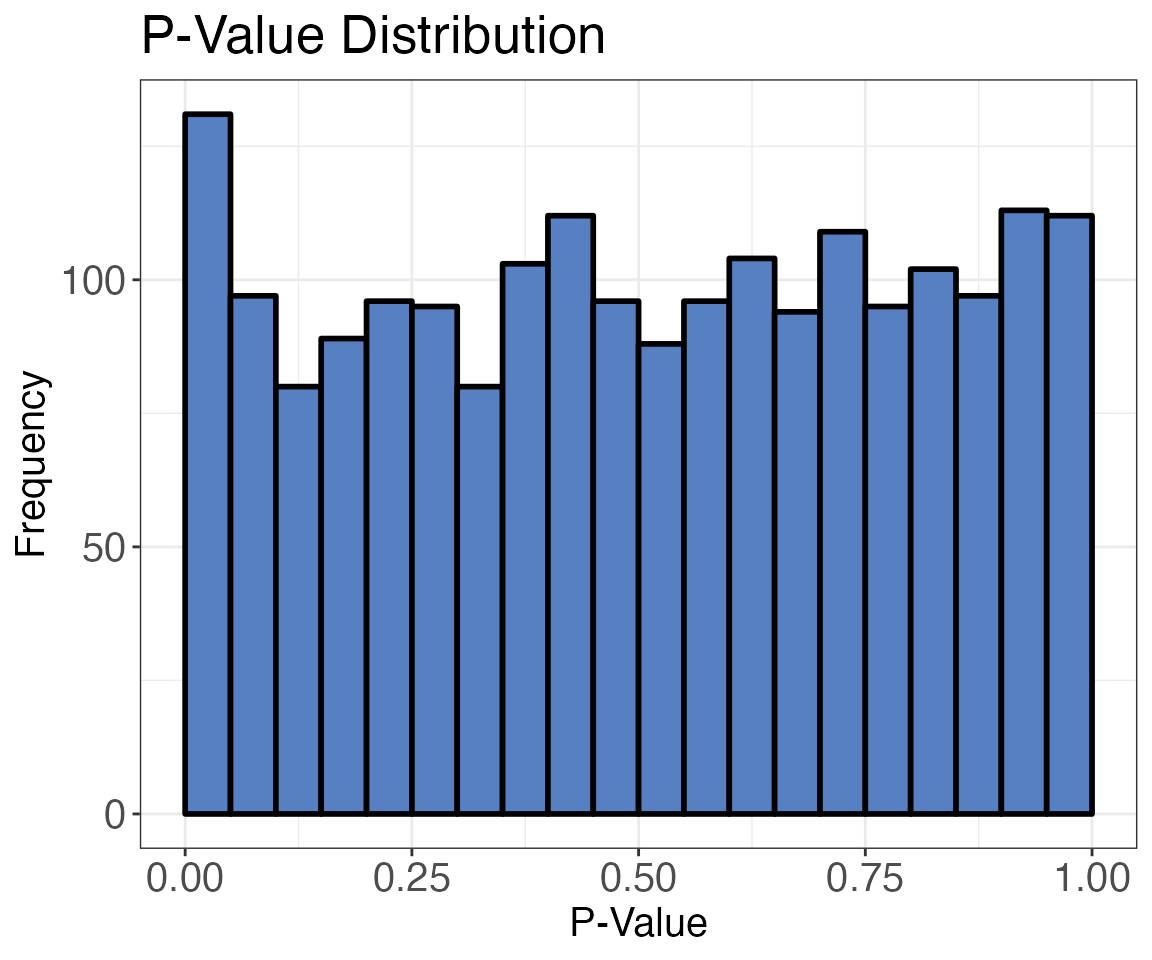
For this subset of data the distribution of p-values is relatively flat and there is no large increase in values in the low p-value range (the distribution is uniform when a lot of your hypotheses are null). This is likely because, for this experiment, only a very small fraction of peptides show changes.
It is recommended to always check the (non-adjusted) p-value distribution. A histogram that does not have a uniform distribution with or without a specific enrichment for very low p-values (due to a treatment induced effect) indicates a failure of the theoretical null distribution, which could have several causes (Efron 2010, chapter 6).
You can read more about p-value distributions here.
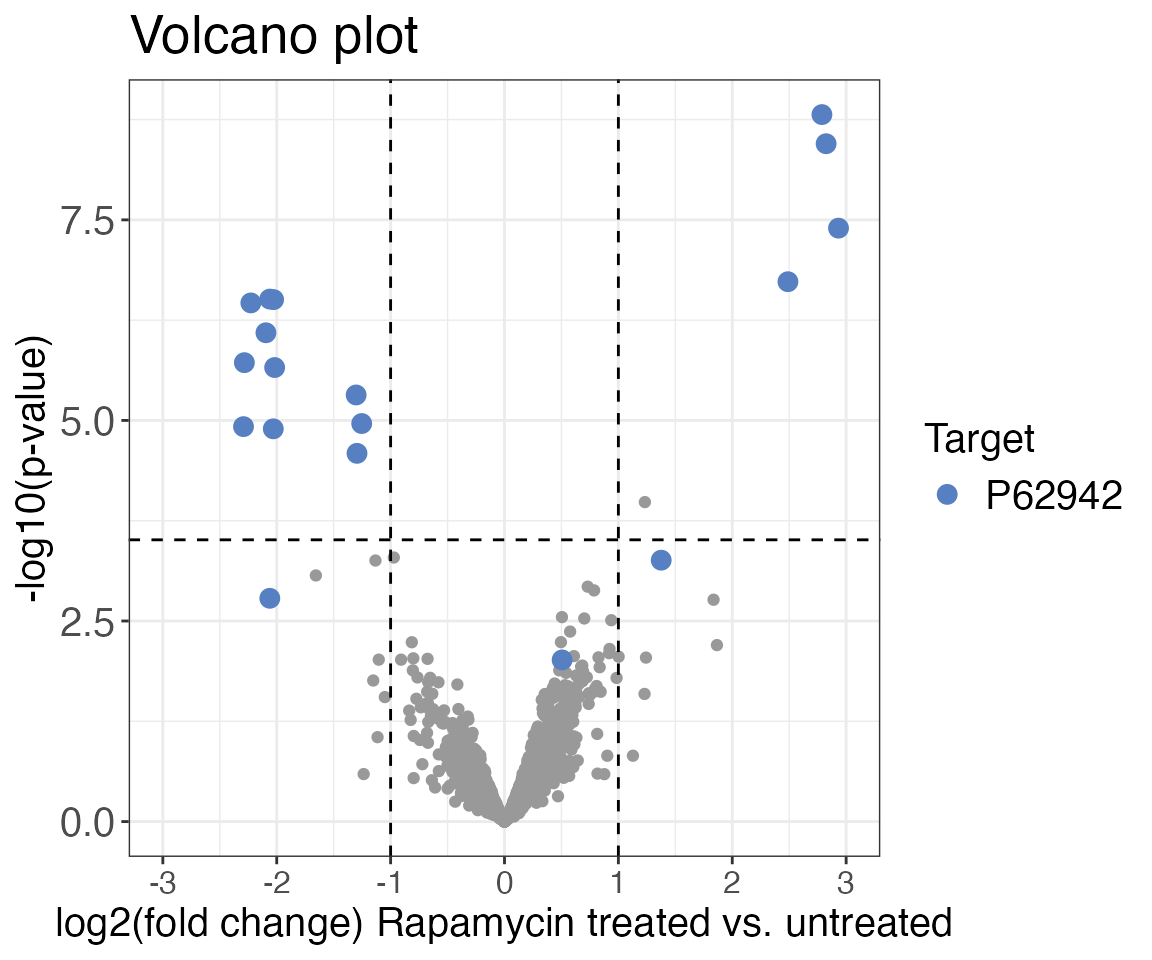
Volcano plot
Next we are going to visualise the output of the previously performed
hypothesis test to assess the results of our experiment. For this we are
going to plot a volcano plot with fold-changes on the x-axis and the
p-value on the y-axis. The output of the previously used
calculate_diff_abundance() function is ideal to use for the
volcano_plot() function as it contains all the information
we need: precursor IDs, protein IDs, fold changes (diff),
p-values (pval) and adjusted p-values
(adj_pval). We are going to highlight the peptides of the
known target of rapamycin FKBP12 (UniProt ID = P62942) in blue to
quickly find the peptides in the plot. You can also make the plot
interactive by setting interactive = TRUE. This will help
you quickly obtain more information on each point in the plot.
Since adjusted p-values are related to unadjusted p-values often in a
complex way, it makes them hard to be interpret if they would be used
for the y-axis. To nevertheless use the information of adjusted p-values
for the plot, you can provide the column name of the adjusted p-values
to the significance_cutoff argument next to the desired
cutoff. The function will look for the closest adjusted p-values above
and below the set cutoff and take the mean of the corresponding p-value
as the cutoff line. If there is no adjusted p-value in the data that is
below the set cutoff no line is displayed. This allows you to display
volcano plots using p-values while using adjusted p-values for the
cutoff criteria.
volcano_plot(
data = diff_abundance_data,
grouping = eg_precursor_id,
log2FC = diff,
significance = pval,
method = "target",
target_column = pg_protein_accessions,
target = "P62942",
x_axis_label = "log2(fold change) Rapamycin treated vs. untreated",
significance_cutoff = c(0.05, "adj_pval")
)
# The significance_cutoff argument can also just be used for a
# regular cutoff line by just providing the cutoff value, e.g.
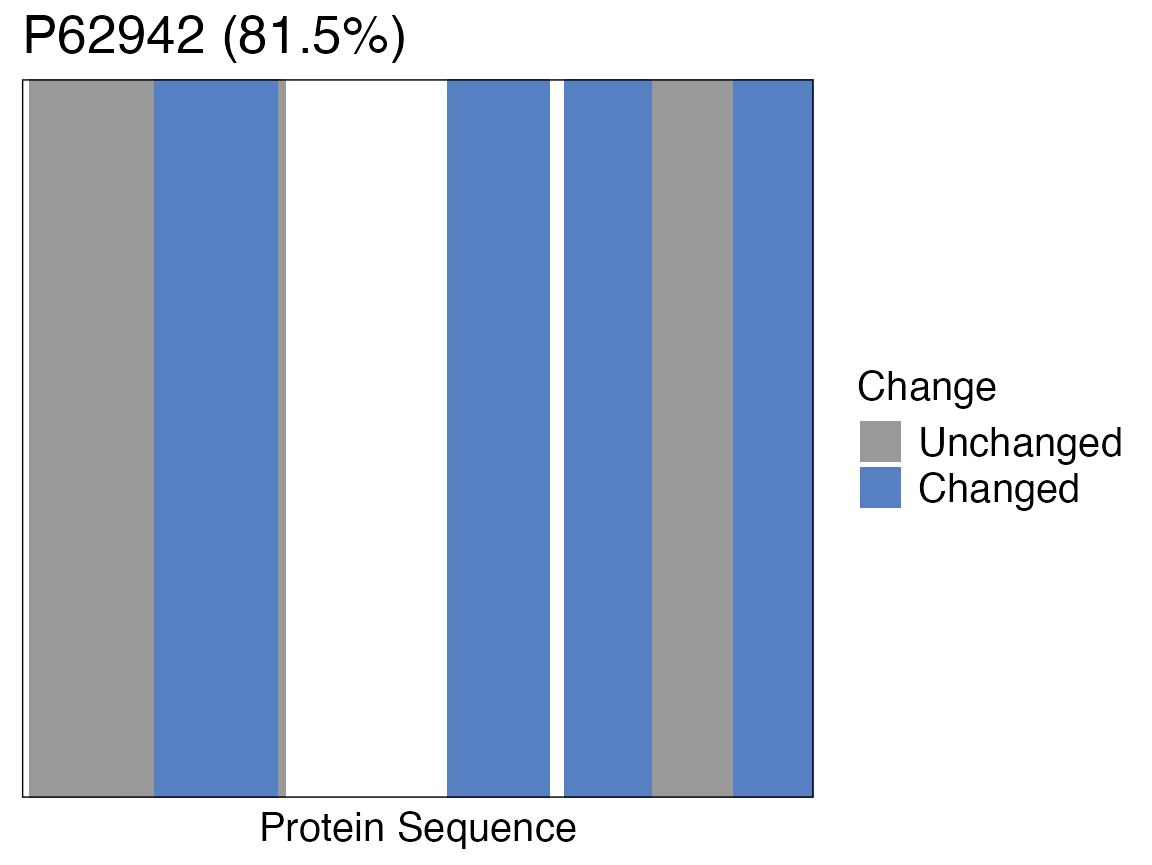
# signficiance_cutoff = 0.05Barcode plot
For LiP-MS experiments a good way to see where on the protein the
changes due to binding or conformational changes occur is to plot a
barcode plot. A barcode plot can be created with the
protti function barcode_plot(). The
detected peptides are coloured in grey and the changing peptides are
highlighted in blue.
In order to produce a barcode plot only for our target FKBP12 we
create a data frame that contains only information for our target
protein using dplyr’s filter() function. The
filtered data frame is then used as the input for the plot.
FKBP12 <- diff_abundance_data %>%
filter(pg_protein_accessions == "P62942")
barcode_plot(
data = FKBP12,
start_position = start,
end_position = end,
protein_length = length,
coverage = coverage,
colouring = diff,
cutoffs = c(diff = 1, adj_pval = 0.05),
protein_id = pg_protein_accessions
)
Wood’s plot
An additional way to plot LiP-MS changes is the Woods’ plot. This plot will show the extent of the precursor fold changes along the protein sequence. The precursors are located on the x-axis based on their start and end positions. The position on the y-axis displays the fold change. The vertical size (y-axis) of the box representing the precurors does not have any meaning.
To produce a Woods’ plot we use the function
woods_plot() and colour the peptides according to their
adjusted p-values. We are highlighting significant adjusted p-values
(< 0.01) with an asterisk. Peptides can also be coloured by another
categorical or continous variable. Asterisks can be added for any
logical (binary) variable.
FKBP12 <- FKBP12 %>%
mutate(significant = ifelse(adj_pval < 0.01, TRUE, FALSE))
woods_plot(
data = FKBP12,
fold_change = diff,
start_position = start,
end_position = end,
protein_length = length,
coverage = coverage,
colouring = adj_pval,
protein_id = pg_protein_accessions,
facet = FALSE,
fold_change_cutoff = 1,
highlight = significant
)
#> $`P62942 (81.5%)`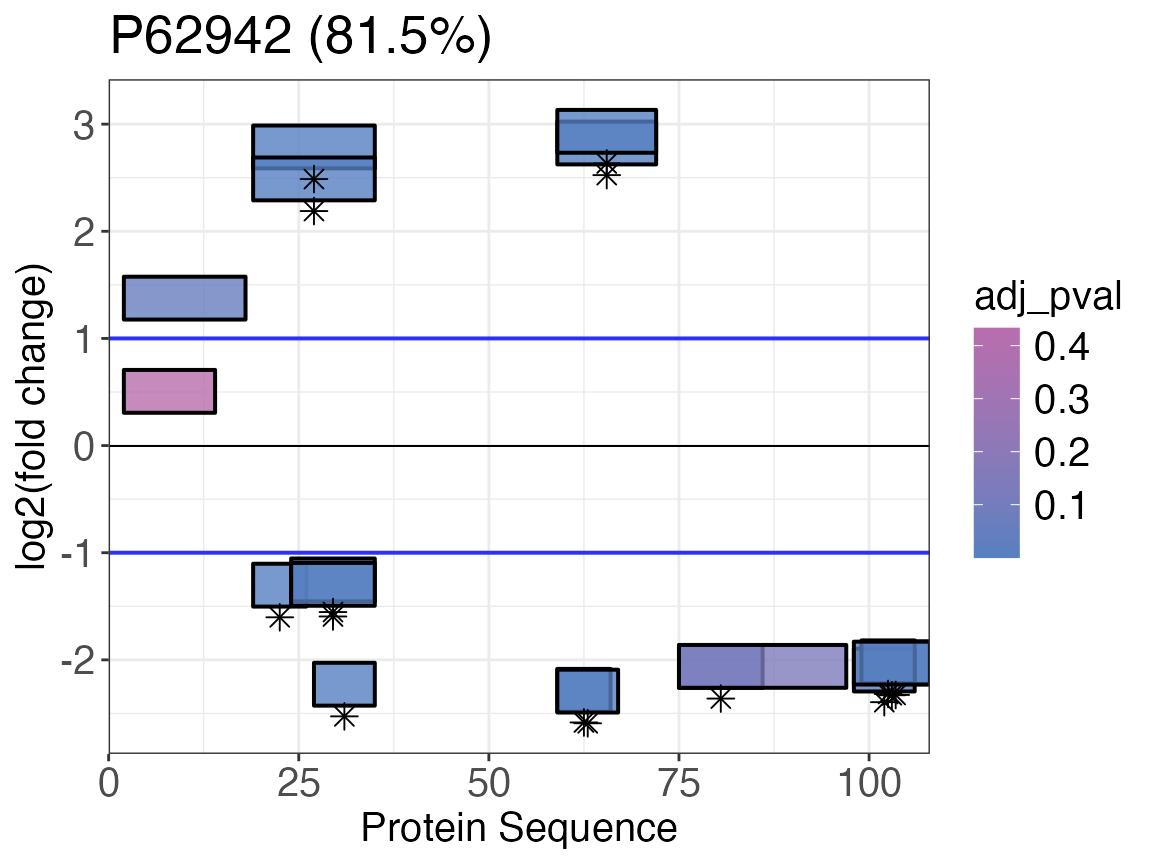
Peptide profile plots
To see how the individual precursors in our target protein are
changing with the treatment we plot profile plots by using the function
peptide_profile_plot(). This is particularly useful as you
can quickly see if your whole protein changes in abundance or only a
fraction of precursors/peptides. If you have protein abundance data you
can also use the plot to show changes in protein abundance over your
treatment condition(s). By selecting multiple targets (as a vector) you
can produce the plot for multiple proteins.
FKBP12_intensity <- data_filtered_uniprot %>%
filter(pg_protein_accessions == "P62942")
peptide_profile_plot(
data = FKBP12_intensity,
sample = r_file_name,
peptide = eg_precursor_id,
intensity_log2 = normalised_intensity_log2,
grouping = pg_protein_accessions,
targets = "P62942",
protein_abundance_plot = FALSE
)
#> $P62942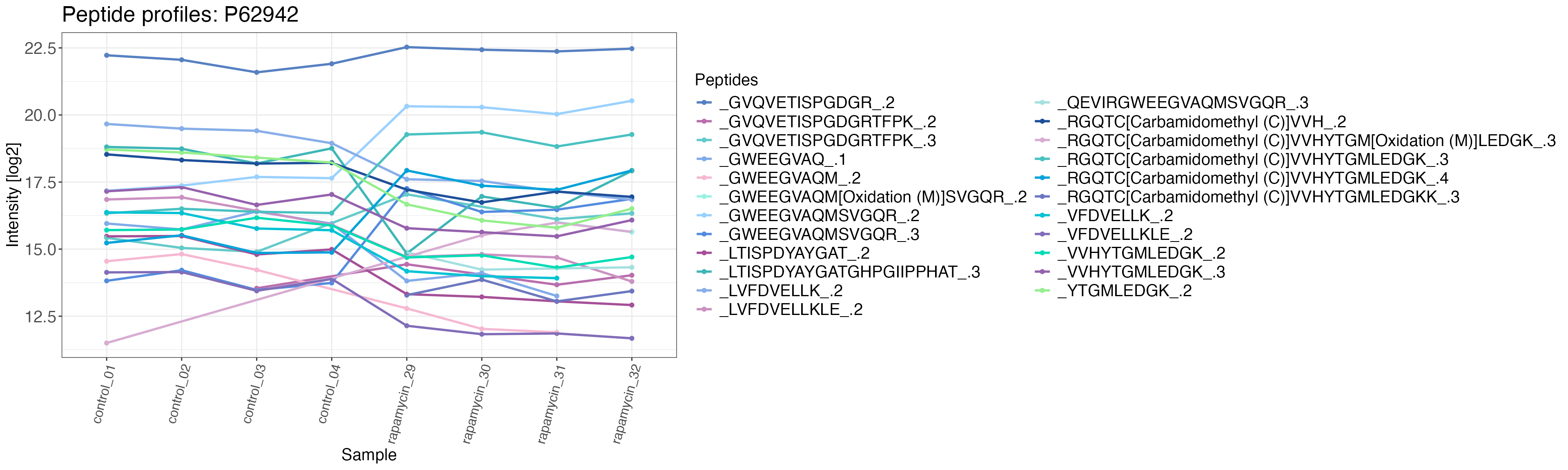
Additional helpful functions
protti includes additional helpful functions that do
not make sense to use for this data set but apply to data sets of full
size that have global changes. These functions include the
calculate_go_enrichment()function that helps you check if
your hits are enriched for specific gene ontology (GO) terms, the
analyse_functional_network() function that plots a String
network based on information from StringDB for your hits and the
calculate_kegg_enrichment() function which checks for
enriched pathways in your hits. Furthermore, you can directly check for
enrichment of a self defined treatment with the
calculate_treatment_enrichment() function.
For GO enrichment you would add an additional column to your data
frame containing information on whether your hit is significant or not.
You can do this by using the dplyr function
mutate(). Here we want the column to contain logicals that
are either TRUE when the adjusted p-value is below 0.05 and the
log2(fold change) is below -1 or above 1 or to be FALSE if this is not
the case. We use the ifelse() function to produce the
logicals. Furthermore, we annotate if the hit is true positive by
marking peptides of the known rapamycin binding protein FKBP12.
For the network analysis we filter the previously produced data frame
containing the is_significant column for significant hits.
This data frame can then be used as an input for
analyse_functional_network() to check if the proteins can
be found in an interaction network based on StringDB information.
For calculate_kegg_enrichment() you need to first use
the function fetch_kegg() to obtain the KEGG pathway
identifiers for your data set. You can then use dplyr’s
right_join()to join the output with the previously produced
data frame containing a column indicating whether your hits are
significant or not.
If you know all known interactors of your specific treatment you can
check for an enrichment of these with the
calculate_treatment_enrichment() function. This is
particularly useful if your treatment has an effect on many
proteins.
diff_abundance_significant <- diff_abundance_data %>%
# mark significant peptides
mutate(is_significant = ifelse((adj_pval < 0.01 & abs(diff) > 1), TRUE, FALSE)) %>%
# mark true positive hits
mutate(binds_treatment = pg_protein_accessions == "P62942")
### GO enrichment using "molecular function" annotation from UniProt
calculate_go_enrichment(
data = diff_abundance_significant,
protein_id = pg_protein_accessions,
is_significant = is_significant,
go_annotations_uniprot = go_f
)
### Network analysis
network_input <- diff_abundance_significant %>%
filter(is_significant == TRUE)
analyse_functional_network(
data = network_input,
protein_id = pg_protein_accessions,
string_id = xref_string,
binds_treatment = binds_treatment,
organism_id = 9606
)
### KEGG pathway enrichment
# First you need to load KEGG pathway annotations from the KEGG database
# for your specific organism of interest. In this case HeLa cells were
# used, therefore the organism of interest is homo sapiens (hsa)
kegg <- fetch_kegg(species = "hsa")
# Next we need to annotate our data with KEGG pathway IDs and perform enrichment analysis
diff_abundance_significant %>%
# columns containing proteins IDs are named differently
left_join(kegg, by = c("pg_protein_accessions" = "uniprot_id")) %>%
calculate_kegg_enrichment(
protein_id = pg_protein_accessions,
is_significant = is_significant,
pathway_id = pathway_id,
pathway_name = pathway_name
)
### Treatment enrichment analysis
calculate_treatment_enrichment(diff_abundance_significant,
protein_id = pg_protein_accessions,
is_significant = is_significant,
binds_treatment = binds_treatment,
treatment_name = "Rapamycin"
)