Calculates and plots ranked intensities for proteins, peptides or precursors.
qc_ranked_intensities(
data,
sample,
grouping,
intensity_log2,
facet = FALSE,
plot = FALSE,
y_axis_transformation = "log10",
interactive = FALSE
)Arguments
- data
a data frame that contains at least sample names, grouping identifiers (precursor, peptide or protein) and log2 transformed intensities for each grouping identifier.
- sample
a character column in the
datadata frame that contains the sample names.- grouping
a character column in the
datadata frame that contains protein, precursor, or peptide identifiers.- intensity_log2
a numeric column in the
datadata frame that contains the log2 transformed intensities of the selected grouping variable.- facet
a logical value that specifies whether the calculation should be done group wise by sample and if the resulting plot should be faceted by sample. (default is
FALSE). Iffacet = FALSEthe median of each protein intensity will be returned.- plot
a logical value that specifies whether the result should be plotted (default is
FALSE).- y_axis_transformation
a character value that determines that y-axis transformation. The value is either "log2" or "log10" (default is "log10").
- interactive
a logical value that specifies whether the plot should be interactive (default is
FALSE).
Value
A data frame containing the ranked intensities is returned. If plot = TRUE a plot
is returned. The intensities are log10 transformed for the plot.
Examples
set.seed(123) # Makes example reproducible
# Create synthetic data
data <- create_synthetic_data(
n_proteins = 50,
frac_change = 0.05,
n_replicates = 4,
n_conditions = 3,
method = "effect_random",
additional_metadata = FALSE
)
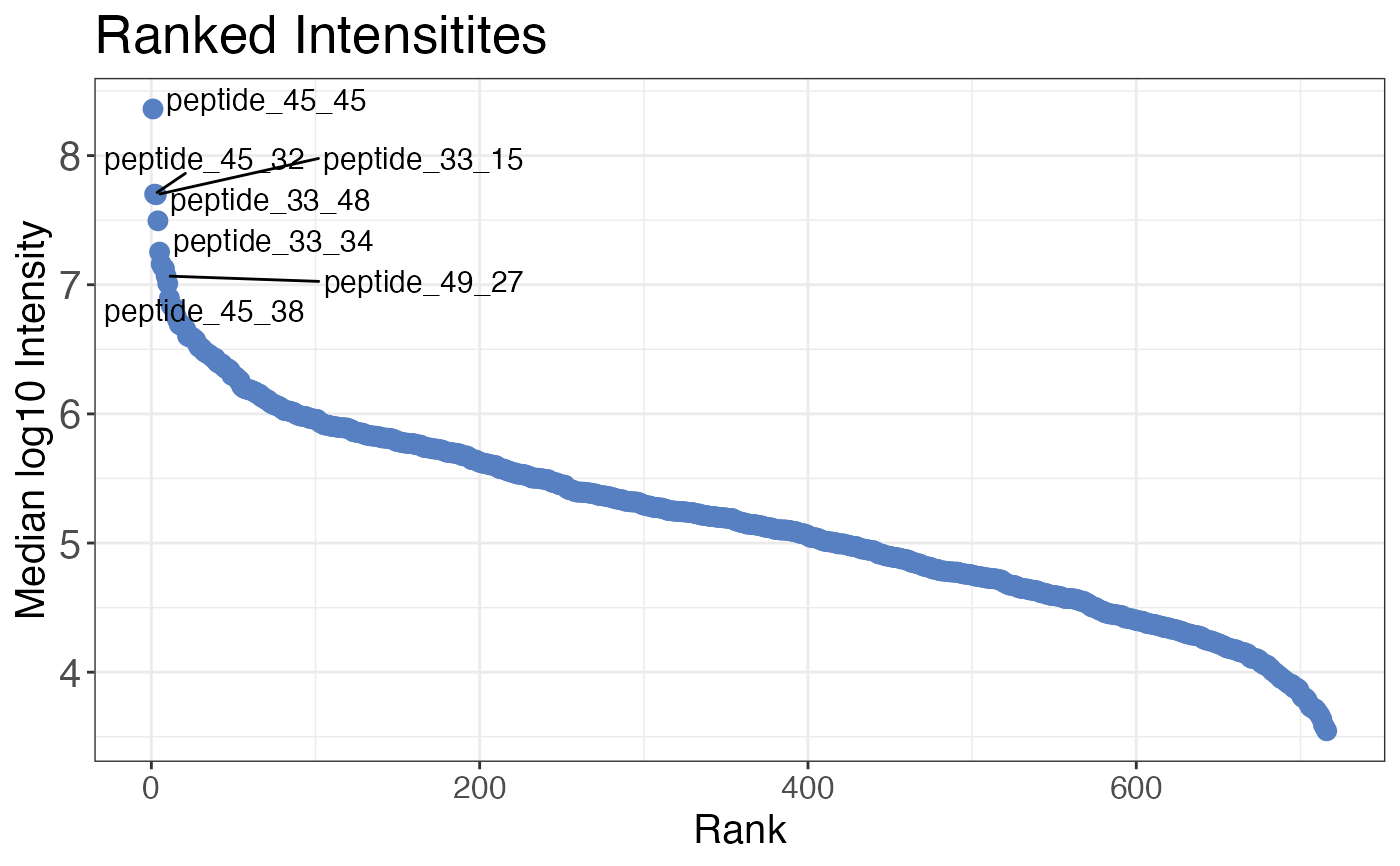
# Plot ranked intensities for all samples combined
qc_ranked_intensities(
data = data,
sample = sample,
grouping = peptide,
intensity_log2 = peptide_intensity,
plot = TRUE,
)
#> Warning: ggrepel: 9 unlabeled data points (too many overlaps). Consider increasing max.overlaps
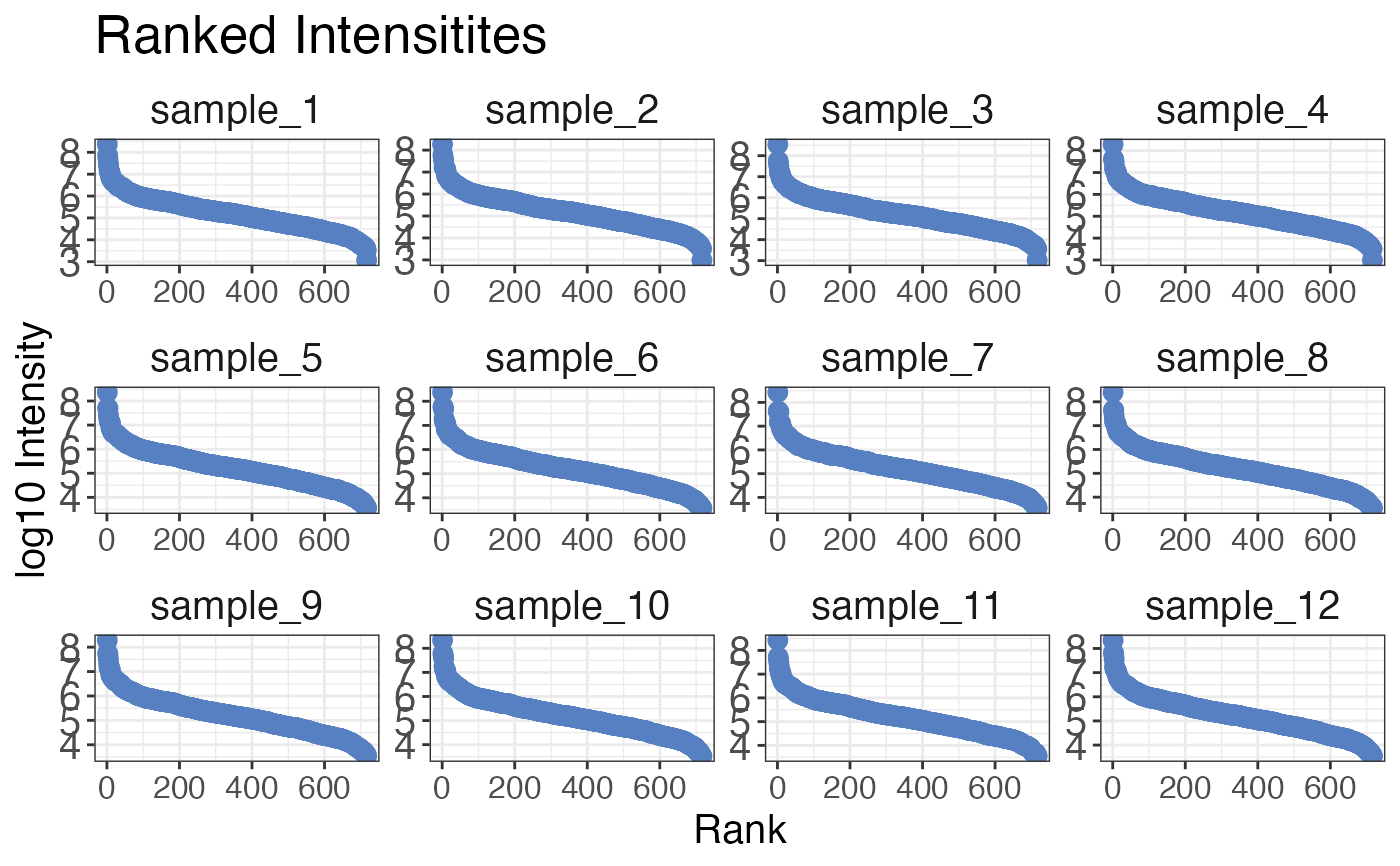
 # Plot ranked intensities for each sample separately
qc_ranked_intensities(
data = data,
sample = sample,
grouping = peptide,
intensity_log2 = peptide_intensity,
plot = TRUE,
facet = TRUE
)
#> Warning: ggrepel: 19 unlabeled data points (too many overlaps). Consider increasing max.overlaps
#> Warning: ggrepel: 19 unlabeled data points (too many overlaps). Consider increasing max.overlaps
#> Warning: ggrepel: 19 unlabeled data points (too many overlaps). Consider increasing max.overlaps
#> Warning: ggrepel: 18 unlabeled data points (too many overlaps). Consider increasing max.overlaps
#> Warning: ggrepel: 19 unlabeled data points (too many overlaps). Consider increasing max.overlaps
#> Warning: ggrepel: 19 unlabeled data points (too many overlaps). Consider increasing max.overlaps
#> Warning: ggrepel: 19 unlabeled data points (too many overlaps). Consider increasing max.overlaps
#> Warning: ggrepel: 19 unlabeled data points (too many overlaps). Consider increasing max.overlaps
#> Warning: ggrepel: 19 unlabeled data points (too many overlaps). Consider increasing max.overlaps
#> Warning: ggrepel: 18 unlabeled data points (too many overlaps). Consider increasing max.overlaps
#> Warning: ggrepel: 19 unlabeled data points (too many overlaps). Consider increasing max.overlaps
#> Warning: ggrepel: 19 unlabeled data points (too many overlaps). Consider increasing max.overlaps
# Plot ranked intensities for each sample separately
qc_ranked_intensities(
data = data,
sample = sample,
grouping = peptide,
intensity_log2 = peptide_intensity,
plot = TRUE,
facet = TRUE
)
#> Warning: ggrepel: 19 unlabeled data points (too many overlaps). Consider increasing max.overlaps
#> Warning: ggrepel: 19 unlabeled data points (too many overlaps). Consider increasing max.overlaps
#> Warning: ggrepel: 19 unlabeled data points (too many overlaps). Consider increasing max.overlaps
#> Warning: ggrepel: 18 unlabeled data points (too many overlaps). Consider increasing max.overlaps
#> Warning: ggrepel: 19 unlabeled data points (too many overlaps). Consider increasing max.overlaps
#> Warning: ggrepel: 19 unlabeled data points (too many overlaps). Consider increasing max.overlaps
#> Warning: ggrepel: 19 unlabeled data points (too many overlaps). Consider increasing max.overlaps
#> Warning: ggrepel: 19 unlabeled data points (too many overlaps). Consider increasing max.overlaps
#> Warning: ggrepel: 19 unlabeled data points (too many overlaps). Consider increasing max.overlaps
#> Warning: ggrepel: 18 unlabeled data points (too many overlaps). Consider increasing max.overlaps
#> Warning: ggrepel: 19 unlabeled data points (too many overlaps). Consider increasing max.overlaps
#> Warning: ggrepel: 19 unlabeled data points (too many overlaps). Consider increasing max.overlaps