Correlation based hirachical clustering of samples
Source:R/qc_sample_correlation.R
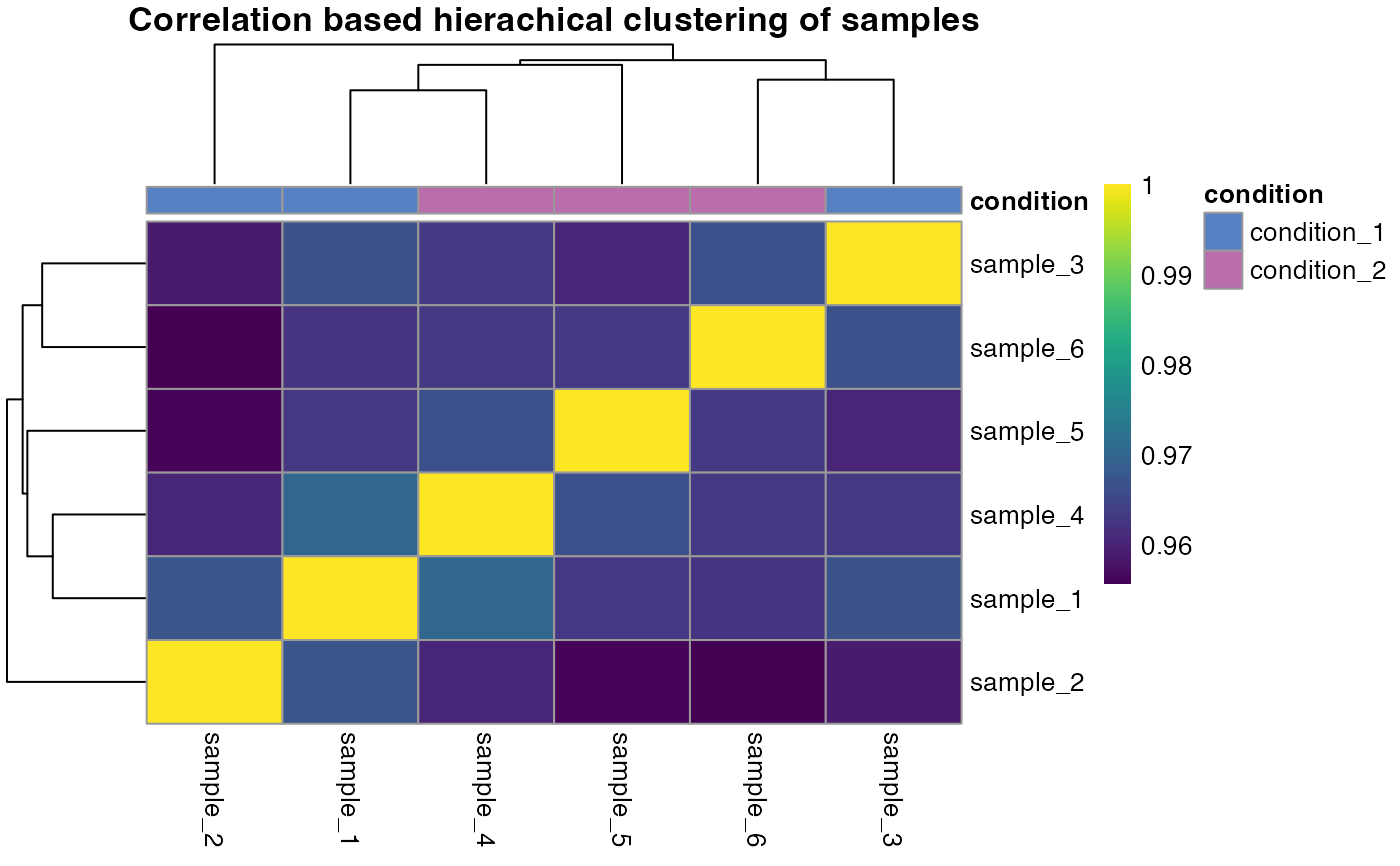
qc_sample_correlation.RdA correlation heatmap is created that uses hirachical clustering to determine sample similarity.
qc_sample_correlation(
data,
sample,
grouping,
intensity_log2,
condition,
digestion = NULL,
run_order = NULL,
method = "spearman",
interactive = FALSE
)Arguments
- data
a data frame that contains at least the input variables.
- sample
a character column in the
datadata frame that contains the sample names.- grouping
a character column in the
datadata frame that contains precursor or peptide identifiers.- intensity_log2
a numeric column in the
datadata frame that contains log2 intensity values.- condition
a character or numeric column in the
datadata frame that contains the conditions.- digestion
optional, a character column in the
datadata frame that contains information about the digestion method used. e.g. "LiP" or "tryptic control".- run_order
optional, a character or numeric column in the
datadata frame that contains the order in which samples were measured. Useful to investigate batch effects due to run order.- method
a character value that specifies the method to be used for correlation.
"spearman"is the default but can be changed to"pearson"or"kendall".- interactive
a logical value that specifies whether the plot should be interactive. Determines if an interactive or static heatmap should be created using
heatmaplyorpheatmap, respectively.
Value
A correlation heatmap that compares each sample. The dendrogram is sorted by optimal leaf ordering.
Examples
# \donttest{
set.seed(123) # Makes example reproducible
# Create example data
data <- create_synthetic_data(
n_proteins = 100,
frac_change = 0.05,
n_replicates = 3,
n_conditions = 2,
method = "effect_random"
)
# Create sample correlation heatmap
qc_sample_correlation(
data = data,
sample = sample,
grouping = peptide,
intensity_log2 = peptide_intensity_missing,
condition = condition
)
 # }
# }